Figures & data
Table 1 Details for GEO data
Figure 1 Log2FC heat map of the top 20 upregulated and downregulated genes of the five expression-microarray groups downloaded from the GEO data set.
Notes: The differentially expressed gene (DEG) list of each expression microarray from the GEO data was integrated using R software with the RRA package. The abscissa is the GEO ID, and the ordinate is the gene name. Red represents upregulated gene expression, and green represents downregulated gene expression in hepatocellular carcinoma tissues compared with adjacent normal liver tissues. Numbers in the box represent log2FC values that resulted from the integrated analysis.
Abbreviations: FC, fold change; GEO, Gene Expression Omnibus; RRA, robust rank aggregation.
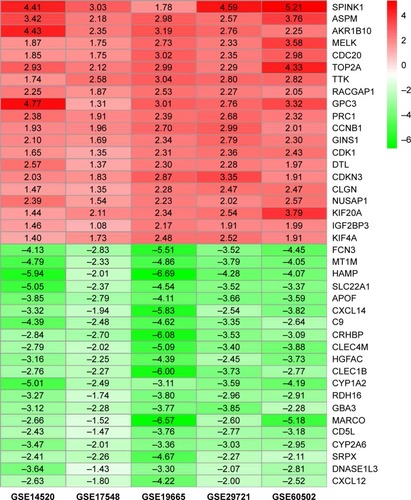
Figure 2 Hierarchical clustering heat map (A) and volcano plot (B) of differentially expressed genes screened based on TCGA HCC WTS data.
Notes: Protein-encoding mRNA-expression data were screened and analyzed using R software and the Limma package. Log2FC >1 and P<0.05 were used as cutoff criteria.
Abbreviations: FC, fold change; TCGA, the Cancer Genome Atlas; HCC, hepatocellular carcinoma; WTS, whole-transcriptome sequencing; FDR, false discovery rate.
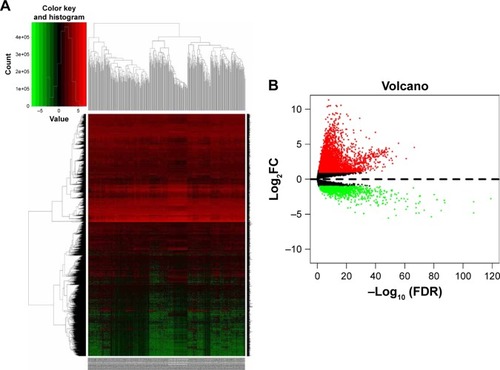
Table 2 Potential prognostic genes in hepatocellular carcinoma
Table 3 KEGG pathway analysis of potential prognostic genes associated with HCC
Figure 3 Hierarchical clustering heat map and volcano plot of screened DEMs (A, B) and DELs (C, D) based on TCGA HCC WTS data.
Notes: miRNA- and lncRNA-expression data were downloaded and analyzed using R software and the Limma package. Log2FC >2 and P<0.01 were used as cutoff criteria.
Abbreviations: DEMs, differentially expressed miRNAs; DELs, differentially expressed lncRNAs; FC, fold change; TCGA, the Cancer Genome Atlas; HCC, hepatocellular carcinoma; WTS, whole-transcriptome sequencing.
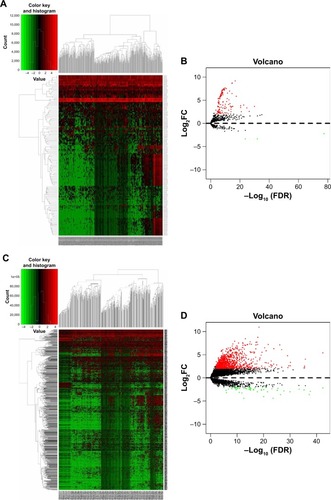
Table 4 Interaction between genes in ceRNA regulatory network
Figure 4 The ceRNA regulatory network in HCC.
Notes: The oval represents mRNA, the diamond represents lncRNA, and the square represents miRNA. Red indicates the gene was upregulated and green indicates the gene downregulated in HCC tissues compared with adjacent normal liver tissues. The line between the genes indicates that there could be regulatory relationships between the two genes.
Abbreviations: ceRNA, the competing endogenous RNA; HCC, hepatocellular carcinoma.
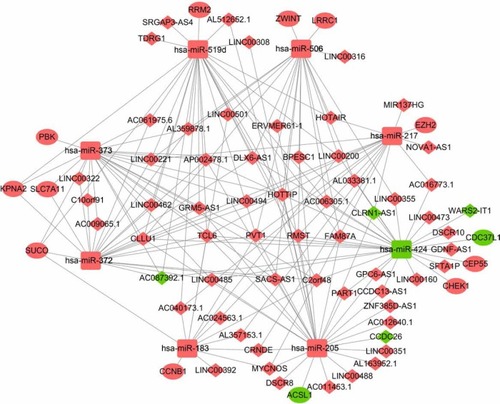
Figure 5 Kaplan–Meier curve analysis was performed on the DELs in the ceRNA regulatory network. P<0.05 was used as the cutoff criterion.
Abbreviations: DELs, differentially expressed lncRNAs; ceRNA, competing endogenous RNA.
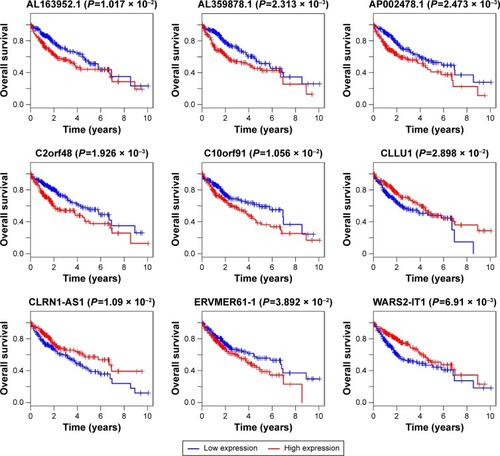
Figure 6 mRNA-expression levels in the ceRNA regulatory network of the 20 pairs of HCC tissue and adjacent normal liver tissue.
Notes: Experiments were repeated three times. *P<0.05.
Abbreviations: ceRNA, competing endogenous RNA; HCC, hepatocellular carcinoma.
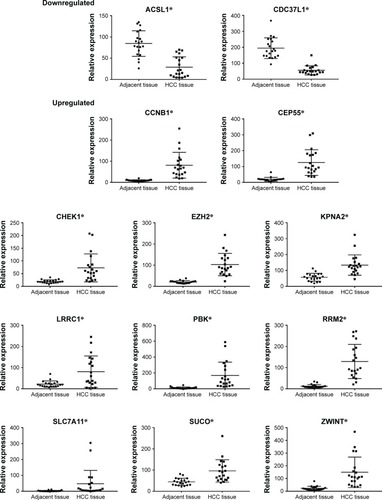
Figure S1 Differential gene expression between HCC tissue and adjacent normal liver tissue from five expression-microarray groups.
Notes: (A) GSE14520 data; (B) GSE17548 data; (C) GSE19665 data; (D) GSE29721 data; (E) GSE60502 data. The red points represent genes that were upregulated and the green points those that were downregulated in HCC tissue compared with adjacent normal liver tissue, which were analyzed using R software and the Limma package. The black points represent genes without a significant difference between the two tissue types. Log2FC >1 and P<0.05 were used as cutoff criteria. Microarray groups downloaded from the GEO data set.
Abbreviations: FC, fold change; GEO, Gene Expression Omnibus; HCC, hepatocellular carcinoma.
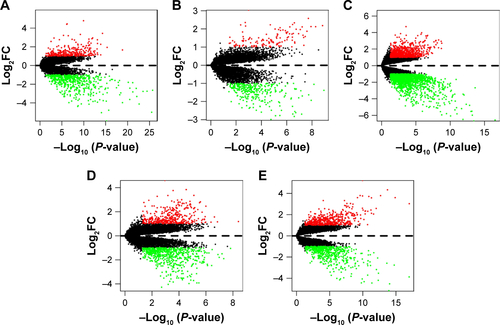
Figure S2 Hierarchical clustering heat maps of differentially expressed genes of five expression-microarray groups, downloaded from the GEO data set.
Notes: (A) GSE14520 data; (B) GSE17548 data; (C) GSE19665 data; (D) GSE29721 data; and (E) GSE60502 data. The color of genes gradually changes with expression-level changes in hepatocellular carcinoma (HCC) tissue compared with adjacent normal liver tissue. Black indicates no significant changes in gene expression. Prominent gene expression in HCC tissue is either upregulated (red color) or downregulated (green color). The depth of color corresponds to expression intensity. Data were analyzed using the R software and the Limma package using. Log2FC >1 and P<0.05 were used as cutoff criteria.
Abbreviations: FC, fold change; GEO, Gene Expression Omnibus.
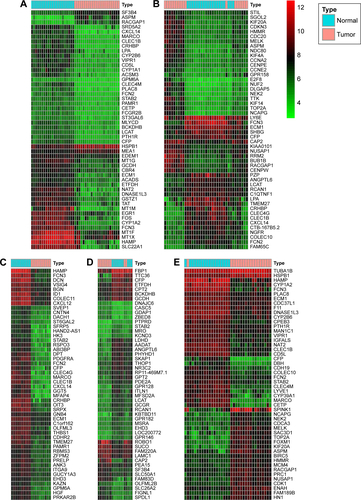
Figure S3 GO enrichment analysis of the potential prognostic genes in hepatocellular carcinoma.
Notes: (A) The GO analysis divided potential prognostic genes into three functional groups: the biological process, cell composition, and molecular function groups. (B) Significant GO enrichment items relating to the potential prognostic genes in the different functional groups.
Abbreviation: GO, Gene Ontology.
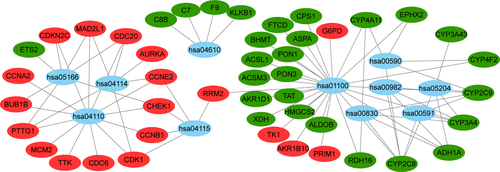
Figure S4 Pathway enrichment of potential prognostic genes.
Note: Blue represents signaling pathways, red represents upregulated genes, and green represents downregulated genes.