Figures & data
Figure 1 Signaling pathways involved in the mechanisms of action of ibrutinib in CLL.
Notes: (A) Antigen binding to the BCR triggers the activation of SYK and BTK. BTK is mostly responsible for the activation of PLCG2. PLCG2 is involved in inducing intracellular calcium release and extracellular calcium influx, followed by the activation of ERK1/ERK2 and NF-κB, as well as NFAT. (B) In parallel, LYN phosphorylates the BCR co-receptor CD19, which activates PI3K. Akt is activated via PI3K. PI3K catalyzes membrane-associated PIP2 to generate PIP3. PIP3 attracts the amino-terminal PH lipid-interaction module of BTK, which in turn allows SYK and LYN to completely activate the BTK enzyme. (C) BTK is essential for CXCR4- and CXCR5-mediated signaling pathways. CXCL12 most probably induces BTK activation through the interaction of heterotrimeric G protein subunits with BTK. (D) Ligands binding to TLR induce the MYD88-mediated activation of NF-κB. BTK has been shown to contribute to TLR signaling by interacting with the intercellular domains of most TLRs.
Abbreviations: ERK1/2, extracellular signal-regulated kinase 1/2 (also termed MAPK3/MAPK1); NF-κB, nuclear factor-κB; PI3K, phosphatidylinositol 3-kinase; NFAT, nuclear factor of activated T cells; PIP2, phosphatidylinositol 4,5-diphosphate; PIP3, phosphatidylinositol 3,4,5-trisphosphate; CXCR4, CXC-chemokine receptor 4; CXCL12, CXC chemokine ligand 12; TLR, Toll-like receptor; MYD88, myeloid differentiation primary response 88.
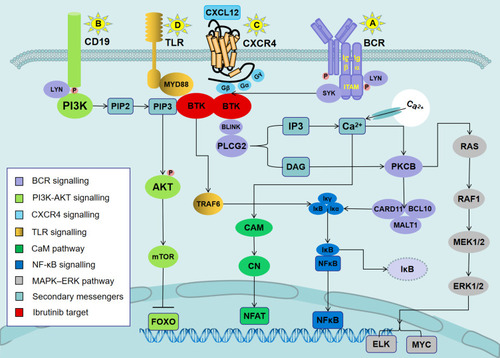
Table 1 Ibrutinib in Previously Untreated CLL Patients
Table 2 Ibrutinib in Previously Treated CLL Patients
Figure 2 Mechanisms of ibrutinib resistance in CLL and mutations in pathways governing BCR signaling.
Notes: (A) The BTK mutation attenuated its binding to ibrutinib, resulting in a reduced ability to inhibit the phosphorylation of downstream molecules, thereby allowing the BCR signal to continue to be passed down. (B) LYN and SYK kinase bypassed BTK to directly activate the mutant PLCG2, which caused increased Ca2+ influx to activate different oncogenic pathways, including PIK3–Akt, NF-κB survival signaling, and the MAP kinase pathway. (C) The binding of IL-4 and IL-6 released from the microenvironment to their corresponding receptors activated JAK kinase, followed by the phosphorylation of the STAT6 or STAT3. The activated STAT upregulated the expression levels of anti-apoptotic proteins MCL-1 and BCL-xL. (D) The acquired short arm of chromosome 2 (2p+) induced the overexpression of XPO1. XPO1 promoted the export of tumor suppressor proteins (such as p53 and FOXO) to the cytoplasm, thereby relieving their inhibitory effect on the cell cycle. (E) Activation of the JAK/STAT signaling pathways might lead to overexpression of PD-L1/PD-L2 in patients with RT.
Abbreviations: IL-4, interleukin 4; JAK, Janus kinase; STAT6, signal transducer and activator of transcription factor 6; MCL-1, myeloid cell leukemia-1; BCL-xL, B-cell lymphoma-xL; XPO1, exportin-1.
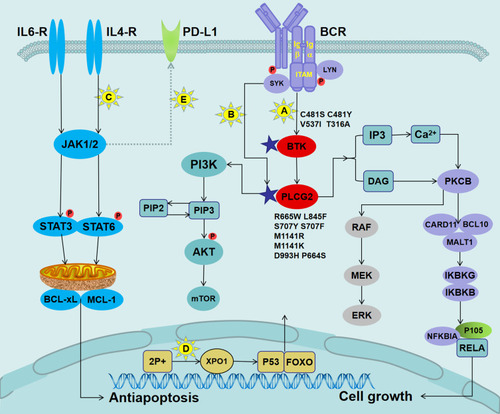
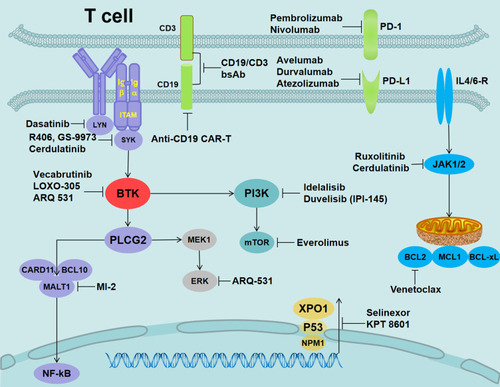
Figure 3 Alternative target inhibitors predicted to overcome ibrutinib resistance.
Notes: LYN inhibition (dasatinib), SYK inhibition (R406, GS-9973, and cerdulatinib), reversible BTKi (vecabrutinib and LOXO-305), nonselective reversible BTKi (ARQ-531), Bcl-2 inhibition (venetoclax), JAK1/2 (ruxolitinib and cerdulatinib), mTOR inhibition (everolimus), MALT1 inhibition (MI-2), XPO1 inhibition (selinexor and KPT 8601), PD-1 inhibition for RT (nivolumab and pembrolizumab), PD-L1 inhibition (avelumab, durvalumab, and atezolizumab), PI3K inhibition (duvelisib/IPI-145 and idelalisib), and CD19 (anti-CD19 CAR-T and CD19/CD3-scFv-Fc bsAb).