Figures & data
Table 1 List of Unarmed oHSVs for GBM Treatment
Table 2 List of Re-Targeted oHSVs for GBM Treatment
Table 3 List of Armed oHSVs for GBM Treatment
Table 4 Clinical Studies with oHSVs in GBM
Figure 1 Strategy to enhance efficacy of oHSV as monotherapy. This figure represents different strategies to enhance the anti-GBM efficacy of oHSV. (A–B) Wild-type HSV can be genetically modified to remove or inactivate various genes to create unarmed oHSV. This is to ensure virus replication selectively occurs in cancer cells, thus enhancing the safety of oHSV (eg, dlsptk, hrR3, 1716, 3616, G207, G47∆, HSVQ1, rQNestin34.5, MG18L). (C) To further enhance oHSV efficacy, different transgene variants are incorporated into oHSV genome to generate armed oHSV. Transgene expression increases oHSV anti-cancer efficacy through enhancement of viral spread (eg, OV-CDH1, OV-ChaseM) or activation of the host immune response (eg, G47∆-mIL12, αMPD-1 scFv) or other means (See ). (D) In addition, insertion of transgene that has similar function to oHSV genes but originating from other viruses is an alternative approach to enhance oHSV replication/oncolysis. oHSVs that are engineered by this method are defined as chimeric oHSV (eg, C130, C134). (E) Wild-type HSV can also be modified to enhance cancer-specificity, based on cellular surface receptor expression profile. They are known as re-targeted oHSVs, which selectively interact with various receptors that exclusively overexpressed in GBM cells but not in normal neurons/or glial cells (eg, R-LM113, KNE). This approach allows maximal oncolysis because HSV genome remains intact. (F) To enhance safety and avoid off-target effects, re-targeted oHSVs (E) can be further genetically modified to include miRNA recognition binding sites (such as miR-124 in KGE-4:T124) whose expression are distinct in GBM cells compared to healthy neurons/glial cells. These recognition-binding sites are incorporated into the untranslated region (UTR) of important genes responsible for viral pathogenicity. Based on the difference between GBM cells and healthy neurons/glial cells, these recognition binding sites can be occupied and the sequential genes are not translated in normal cells, whereas the recognition binding sites are free and viral translation takes place in GBM cells. In addition, re-targeted oHSVs (E) can be armed with different transgene of interest (G) to further enhance anti-tumor efficacy (eg, R-115, KMMP9).
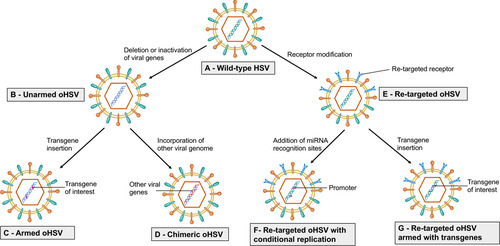
Figure 2 Armed oHSVs for GBM treatment. This figure represents different transgene variants that are inserted in the oHSV genome to enhance the anti-tumor efficacy of oHSV. (A) oHSV armed with cytokine such as IL-12, which induces Th1 differentiation, stimulates growth and cytotoxicity of NKs, increases IFN-γ production, and inhibits angiogenesis. (B) oHSV armed with angiogenic inhibitors decrease tumor vascularity (CD31+ vessels) or VEGF, inhibit anti-viral macrophages, and increase viral spread. (C) oHSV can be engineered to replace one copy of γ34.5 with GADD34. GADD34 binds to PP1 and promotes eIF-2α dephosphorylation - a function that corresponds to γ34.5 in HSV. Since GADD34 does not possess beclin-1-binding motifs of γ34.5, it does not produce neurotoxicity as wild-type HSV containing both copies of γ34.5. (D) Localized expression of anti-PD-1 by an oHSV inhibits PD-1/PD-L1 engagement, prevents T cell exhaustion and unleashes anti-tumor immunity. (E) oHSV can be armed with CDH-1 gene, which encodes for E-cadherin – an adhesion molecule and a ligand for an inhibitory receptor expressed on NK cells (KLRG1). E-cadherin can cooperate with nectin-1 and promote cell-to-cell adherent junctions and enhance cell-to-cell oHSV spread. Another strategy to increase viral spread is to create an oHSV that expresses chondroitinase ABC, which removes side chain of CSPG, prevents extracellular space tortuosity, and thus, facilitates viral spread. (F) PTENα expressed by an oHSV metabolizes PIP3 prevents activation of the PI3K/AKT/mTOR signaling pathway and inhibits tumor growth and migration. (G) oHSV armed with ULBP3 enhances anti-tumor immunity. ULBP3 is a ligand for NKG2D. ULBP3-NKG2D interaction augments the anti-tumor activity of NK cells. (H) oHSV can be armed with pro-drug activating genes such as CYP2B1 (which converts CPA to PM) and shiCE (which converts CPT11 to SN-38) to enhance the conversion of these pro-drugs into their anti-cancer active metabolites.
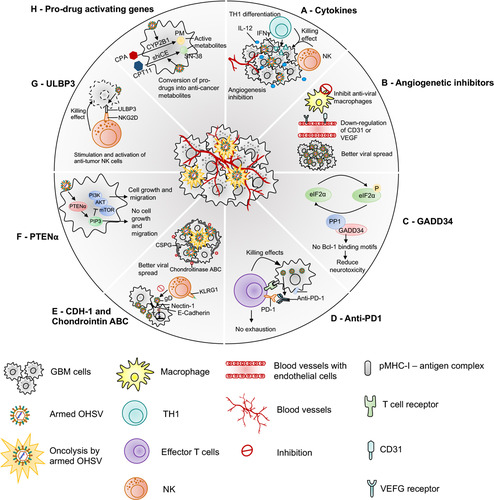
Figure 3 oHSV-based combination therapies for GBM. This figure represents strategies to combine oHSV with different anti-cancer therapies for GBM treatment. (A) oHSV can be synergistically combined with TMZ, a DNA-alkylating agent and an immunomodulator, to induce DNA damage response in MGMT-negative GBM cells. (B) oHSV replication promotes degradation of Rad51 and Chk1 whose functions are important for SSB repair mechanism. Inhibition of PARP in DNA-damaged cells facilitates the conversion of SSB to DSB. The combination of oHSV and PAPRi synergistically induces cell cycle arrest in GBM. (C) HDAC is an enzyme that controls cancer cell survival/progression and upregulates IFN genes. Treatment with HDAC inhibitors prior to oHSV infection inhibits induction of anti-viral IFN genes, resulting in increased transcription of viral genes and improved virus replication. (D) Bortezomib is a proteasome inhibitor, which induces unfolded protein response (UPR), characterized by induction of heat-shock proteins (HSP) 40, 70 and 90, and ER stress in cancercells. Bortezomib-induced ER stress and UPR significantly enhance oHSV replication and synergistic killing of GBM cells. (E) TGF-β plays a critical role in GBM pathogenesis and in maintaining the stemness of GSCs. In TMZ-resistant GSC model, the combination of oHSV and TGF-βi synergistically kills TMZ-resistant recurrent GSCs, increases oHSV replication and induces JNK-MAPK signaling blockade, and eventually inhibits tumor progression. (F) oHSV infection of tumor cells leads to activation of notch signaling in adjacent non-infected tumor cells. Notch signaling pathway plays a critical role in cell-cell interaction and viral spread. Inhibition of notch signaling pathway such as GSI results in increased killing of GBM cells after oHSV therapy. (G) ITGB1, also referred as CD29, plays a critical role in tumor cell proliferation and progression. OS2966 (a humanized CD29 blocking antibody) blocks CD29, reduces the expression of anti-viral genes (IFNα, IFNβ, Stat1, OAS1, OAS2, IRF3, IRF9, and PKR), suppresses oHSV-induced macrophage activation, resulting in enhanced oHSV replication and oncolysis. (H) GBM cells that are infected by oHSV upregulate HMGB1. HMGB1 causes upregulation of ICAM and VCAM, increases vascular permeability and PMBC infiltration to the tumor, leading to edema that might cause CNS injuries. Thus, combination of anti-HMGB1 and oHSV increases survival by reducing brain injuries. (I) Recruitment of NKs after oHSV administration can limit oHSV replication and oHSV-mediated anti-tumor efficacy. Transient inhibition of anti-viral effects of NKs by TGF-β inhibitors enhances viral replication and viral yield. (J) Similarly, transient blockade of TNFα, produced from anti-viral macrophages, by TNFα blocking antibodies or inhibition of STAT1/3 phosphorylation by C16 enhances oHSV replication. In addition, virus-infected cells upregulate CCN1, which in turn activates an intracellular type I IFN response and increases infiltration of macrophages to the site of infection. Treatment with anti-CCN1 reduces virus clearance by macrophages, resulting in better anti-tumor efficacy. (K) Administration of immune checkpoint blockade such as anti-PD-1, anti-PD-L1, anti-CTLA4 prevents T cell exhaustion and enhances oHSV-mediated-anti-tumor immunity. (L) Bevacizumab binds to VEGF, reduces tumor vascularization, decreases vascular permeability, and inhibits tumor growth; however, it also induces tumor cell invasion. An oHSV expressing anti-angiogenic vasculostatin that contains an integrin-antagonizing RGD (Arg-Gly-Asp) motif significantly inhibits glioma cell migration/invasion following bevacizumab treatment, leading to a significant extension of survival compared to bevacizumab monotherapy.
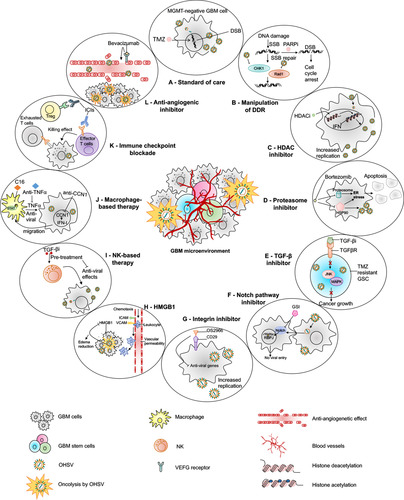
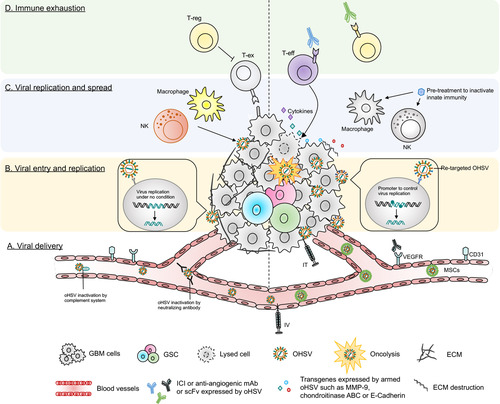
Figure 4 Barriers of current oHSV therapy in GBM and their potential solutions. This figure represents various obstacles of oHSV therapy in GBM (left side) and proposed solutions to overcome these hurdles (right side). (A) Viral delivery – oHSV can be delivered systematically (IV) or locally (IT). IT injection can cause backflow of virus solution to the needle or catheter, leading to insufficient viral dose. IV can be another alternative route to deliver oHSV. Systemic administration of naked OVs can be inactivated by host serum/complement and other immune factors. Systemically delivered oHSVs can be protected from serum/immune factors if delivered by carrier cells, such as MSCs. (B) Viral entry and replication – The higher the expression of HSV’s entry surface receptors such as nectin-1, the higher the entry of oHSVs (such as G207, M032) to cancer cells. Genomic modification attenuates virus replication (such as G207) in tumor cells, especially in GSCs. In contrast, receptor (EGFR, HER2, etc.) re-targeting enhances oHSV entry/replication in cancer cells overexpressing these receptors; however, receptor re-targeting can produce off-target effects in healthy cells that also express the same receptors. This issue can be overcome by incorporating miRNA recognition binding sites (such as miR-124 in KGE-4:T124) whose expression are distinct in GBM cells compared to healthy neurons/glial cells. (C) Viral replication and spread – Condensed ECM can limit viral spread in the TME. oHSVs can be engineered to express E-cadherin, chondroitinase ABC, and/or MMP-9 to destroy the ECM components and facilitate viral spread. In addition, anti-viral innate immunity can also limit viral replication/spread in the TME, as characterized by recruitment of macrophage or NKs that clear virus or virus-infected cells. Transient inhibition of anti-viral innate immune response by pre-treatment with different therapies such as TNF-α or TGF-β inhibitors can help to increase oHSV replication/spread. (D) Immune exhaustion – Successful viral delivery, entry, replication and spread will eventually activate the host’s adaptive anti-tumor immunity, leading to recruitment of T cells to the TME. The recruited T cells oftentimes fail to produce anti-cancer effects due to the expression of T cell exhaustion markers. The presence of Tregs and angiogenesis also contribute to GBM immunosuppression. oHSVs can be combined with systemic ICI or systemic anti-angiogenic mAb, or oHSVs can be engineered to locally express scFv of ICI or anti-angiogenic mAb that should overcome GBM immunosuppression. In addition, oHSVs can be armed with different transgene variants (such as cytokines, co-stimulatory ligands) and testing them in combination with ICIs or other immunotherapies such as anti-cancer vaccines (eg, DCs loaded with tumor-associated antigens) to improve anti-tumor efficacy.