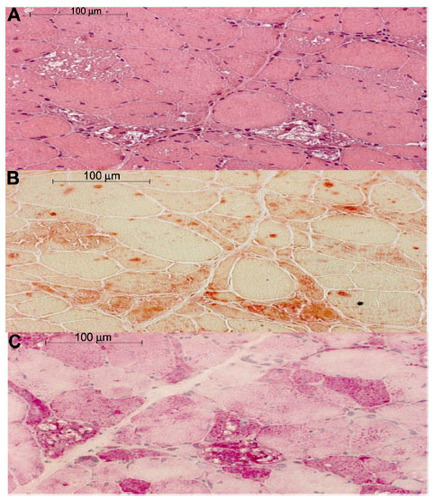
Figures & data
Table 1 Clinical features of classic and non-classic Pompe patients
Table 2 Efficacy of ERT in infantile Pompe disease – study summary
Table 3 Efficacy of ERT in non-classic Pompe disease – study summary
Table 4 Adverse events (AE) and severe adverse events (SAE) reported under ERT