Figures & data
Table 1 Progranulin (GRN)-Related Genes Associated Directly or Indirectly with Frontotemporal Dementia (FTD) and Inherited White-Matter Disorders
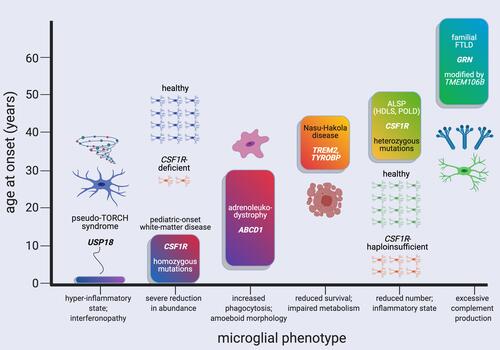
Figure 1 Distinct pathogenic mutations and microglial phenotypes are associated with white-matter disorders with highly variable ages of neurological symptom onset. White-matter diseases and the major microglial phenotypes they may be associated with are ordered according to their typical, approximate age range of onset. The characteristic microglial phenotypes listed are from histopathological studies and/or relevant mouse models of disease; see main text for references. Ages of neurological symptom onset can range from prenatal for type I interferonopathy associated with USP18 deficiency and congenital absence (or near-absence) of microglia due to homozygous mutations in CSF1R, up to the 50s–70s for some cases of frontotemporal lobar degeneration with white-matter hyperintensities associated with pathogenic GRN mutations. Created with BioRender.com.