Figures & data
Table 1 Main Characteristics of the Premature-Ageing Syndromes Described in the Article
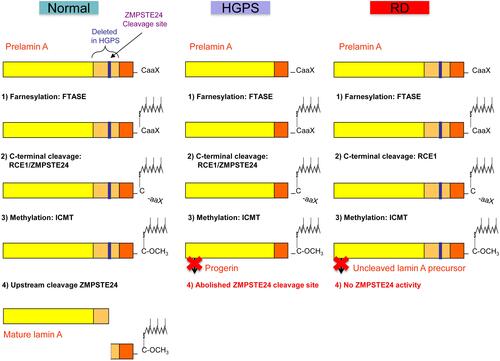
Figure 1 Schematic representation of prelamin A processing in normal individuals, and in patients with Hutchinson–Gilford progeria syndrome and restrictive dermopathy. Notes: Adapted from Coppedè F. The epidemiology of premature aging and associated comorbidities. Clin Interv Aging. 2013;8:1023–1032.Citation5© 2013 Coppedè. Creative Commons Attribution – Non Commercial (unported, v3.0) License (http://creativecommons.org/licenses/by-nc/3.0/). In cells from healthy individuals, prelamin A undergoes a four-step process to become a mature lamin A, including (1) farnesylation of the cysteine residue on the C-terminal CaaX motif by a farnesyltransferase (FTASE), (2) cleavage of the –aaX terminal residues that can be performed either by RCE1 (Ras converting enzyme 1) or by ZMPSTE24 (Zinc metalloprotease related to Ste24p), (3) carboxymethylation of the C-terminal cysteine residue by ICMT (isoprenylcysteine carboxyl methyltransferase), (4) removal of the C‑terminal 15 residues through cleavage by ZMPSTE24. In Hutchinson–Gilford progeria syndrome (HGPS) the common LMNA (p.G608G) mutation results in the deletion of a 50aa region from prelamin A (in orange) containing the upstream ZMPSTE24 cleavage site, resulting in a shorter farnesylated protein that cannot undergo the last maturation step, termed progerin. In restrictive dermopathy (RD) recessive null mutations of ZMPSTE24 result in lack of ZMPSTE24 activity with subsequent accumulation of uncleaved and farnesylated lamin A precursors.