Figures & data
Figure 1 Paroxysmal nocturnal hemoglobinuria (PNH) pathophysiology. A. PIG-A mutations: mutation occurring in the PIG-A gene may affect the synthesis or the function of the encoded protein, resulting in the impairment of the first step of GPI-anchor biosynthesis. As a result, all proteins linked to the cellular membrane through this mechanism are lacking from the surface. B. The dual pathophysiology theory: PIG-A gene mutation may occur without provoking the PNH disease. To induce the clonal expansion necessary for developing clinical PNH, additional factors should occur, which create conditions permissive to expansion of mutated cells. Such extrinsic factors are postulated similar to those inducing stem cell exhaustion in aplastic anemia, which include immune mediated attack of hematopoiesis.
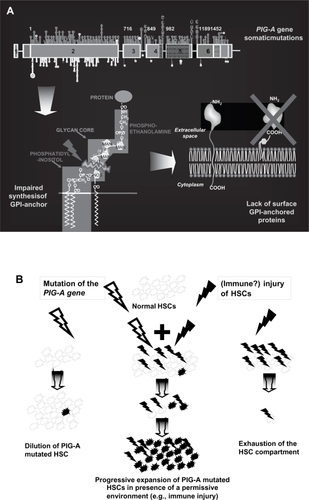
Figure 2 Effect of glycosyl-phosphatidyl inositol anchored proteins (GPI-AP) deficiency on blood cell populations. A. The lack of the complement regulators CD59 and CD55 on red blood cells (RBCs) accounts for their susceptibility to complement mediated lysis resulting in the clinical hallmark of PNH, intravascular hemolysis. B. Deficiency of complement regulators on platelets may account for their putative propensity to activation and aggregation, possibly leading to increased clot formation and thromboembolism. Thrombophilia in PNH may also be related to NO consumption occurring in the presence of increased free Hb resulting from hemolysis, as well as to the impairment of the fibrinolytic system due to the lack of membrane uPAR (see text). C. The lack of anchored proteins GPI-AP has been postulated as putative cause of the expansion of the PNH clone over normal hematopoiesis; the presence of a GPI-linked immune target or a more generic reduced sensitivity to immune effector mechanisms has been hypothesized to explain the immune escape of the PNH clone, but definitive evidence is still lacking.
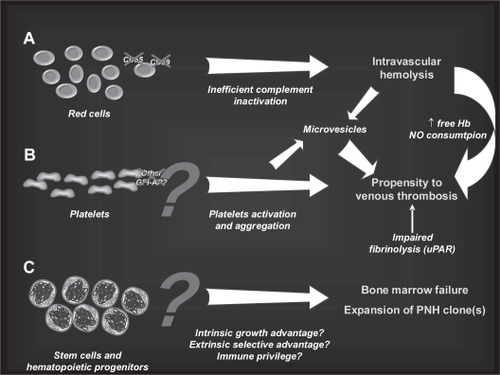
Figure 3 Mechanism of hemolysis in paroxysmal nocturnal hemoglobinuria red blood cells (PNH RBCs). The activation of both the alternative and the classical pathways of the complement cascade converge on the activation of a C5 convertase; PNH RBCs lack CD59, which normally inhibits the assembly of membrane attack complex (MAC), and undergo hemolysis. In addition, the early alternative pathway is dysregulated on PNH as a result of the absence of the C3 convertase inhibitor CD55; thus, PNH RBCs experience uncontrolled complement activation through the alternative pathway. This may explain C3 accumulation on PNH RBCs when hemolysis is blocked in presence of the terminal complement inhibitor eculizumab.
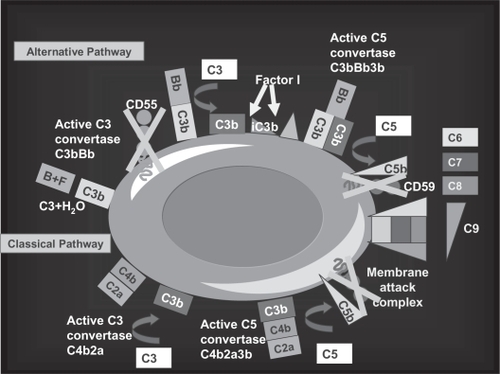
Table 1 Glycosyl-phosphatidyl inositol (GPI)-linked proteins on blood cells and their function
Figure 4 Eculizumab: the molecule. The structure of the anti-complement 5 humanized monoclonal antibody eculizumab (h5G1.1 mAb).
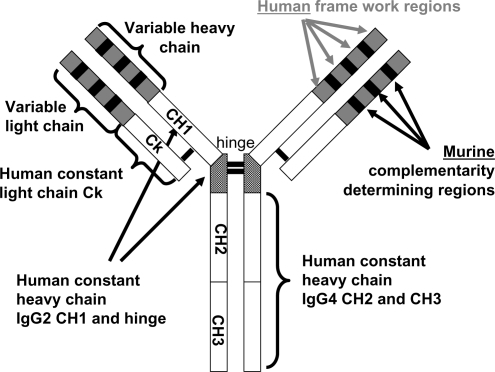
Figure 5 The complement cascade and the C5 blockade by eculizumab. Eculizumab blocks the cleavage of C5 to C5a and C5b; all earlier steps of the complement cascade are preserved, including C3 cleavage.
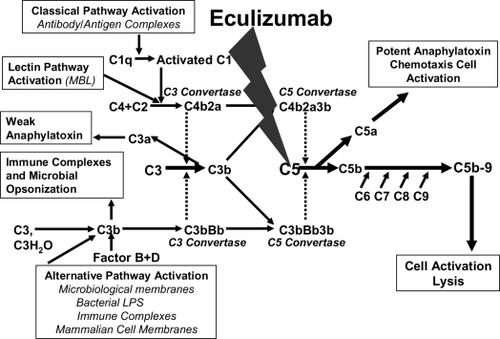
Figure 6 The rationale for eculizumab in paroxysmal nocturnal hemoglobinuria (PNH). A. Normal red blood cells (RBCs) are protected from complement attack by CD59. B. PNH RBCs are susceptible to complement attack due to lack of CD59, resulting in hemolysis and consequent clinical manifestations. C. Eculizumab blocks the complement cascade inhibiting MAC formation; thus, even upon complement activation, PNH RBCs are protected from hemolysis, thus resulting in hemoglobin stabilization and reduction of thromboembolisms.
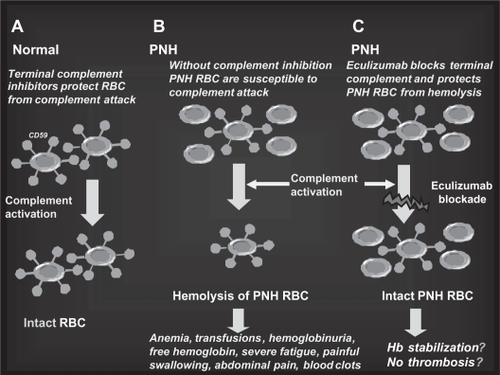
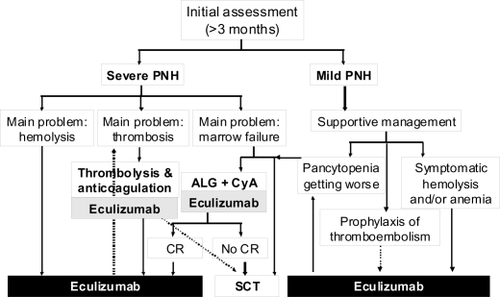
Figure 7 Algorithm of the clinical management of paroxysmal nocturnal hemoglobinuria (PNH) in the eculizumab era. Once PNH is confirmed, all patients suffering from its severe form should receive eculizumab to control intravascular hemolysis; in case of concomitant thromboembolic manifestations an appropriate anticoagulant and/or antithrombotic therapy should be added. In the presence of concomitant severe marrow failure, the indication of an immunosuppressive regimen should be discussed, and programmed possibly before eculizumab treatment (no data on concomitant therapy are available so far). Patients suffering from mild PNH should receive supportive therapy according to the main clinical manifestations: symptomatic hemolysis and/or anemia should be treated by eculizumab, signs of marrow failure may require immunosuppression. Even in this subset the use of eculizumab is reasonable as prophylaxis of thromboembolic events. Indications to stem cell transplantation remain severe marrow failure not responding to immunosuppression (or even as first line in young patients, if a low risk transplant procedure is possible) and possibly refractory thromboembolic disease (but the risk-benefit ratio in comparison to eculizumab has to be demonstrated in this setting).