
Figures & data
Subcutaneous treatment was administered every 8 weeks and oral treatment was administered daily at a dosage of 200 mg/day (100 mg twice daily). Prior to receiving treatment at week 16, patients must have had a ≥30% reduction from baseline in LBPI score at week 16 and a ≥15% reduction from baseline in LBPI score at any week from week 1 to week 15 to continue the study.
LBPI: Low back pain intensity; SC: Subcutaneous.
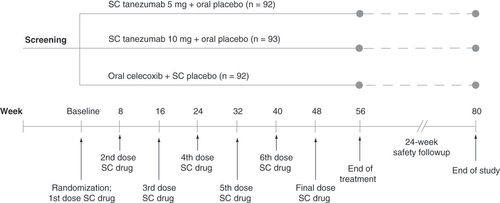
Table 1. Concomitant medication protocols.
The diagram shows the flow of patients throughout the trial. Data are number (%) of patients.
*Two patients were screened but withdrew consent prior to randomization.
†If a patient withdrew due to efficacy reasons other than the specific criteria at week 16 (see Results section), the reason was classified as ‘insufficient clinical response’.
‡Patients completed the study if they completed the safety follow-up period, regardless of whether they completed the treatment phase.

Table 2. Patient demographics and baseline characteristics.
Table 3. Summary of all-causality TEAEs through week 56 (treatment period).
Table 4. Summary of TEAEs of abnormal peripheral sensation through week 56 (treatment period).
Table 5. Summary of TEAEs of possible decreased sympathetic function through week 56 (treatment period).
Table 6. Summary of joint safety through week 80 (treatment + follow-up period).
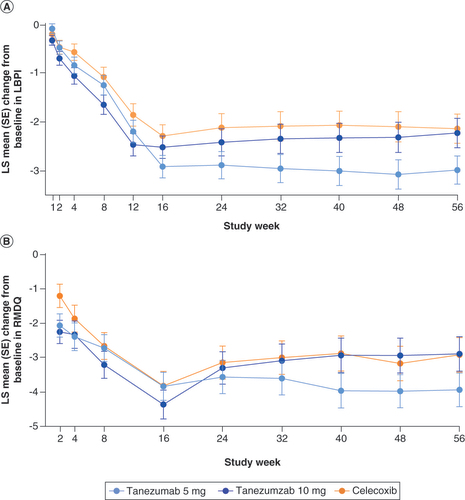
The graphs show LS mean change from baseline in (A) LBPI and (B) RMDQ scores throughout the trial. No formal statistical hypothesis testing was performed.
LBPI: Low back pain intensity; LS: Least squares; RMDQ: Roland Morris disability questionnaire; SE: Standard error.
Supplemental Document 1
Download MS Word (21.8 KB)Supplemental Document 2
Download MS Word (34.4 KB)Supplemental Document 3
Download MS Word (25.8 KB)Supplemental Document 4
Download MS Word (27 KB)Data sharing statement
The authors certify that this manuscript reports original clinical trial data, NCT02725411. Upon request, and patient to certain criteria, conditions and exceptions (see https://www.pfizer.com/science/clinical-trials/trial-data-and-results for more information), Pfizer will provide access to individual de-identified participant data from Pfizer-sponsored global interventional clinical studies conducted for medicines, vaccines and medical devices (1) for indications that have been approved in the US and/or EU or (2) in programs that have been terminated (i.e., development for all indications has been discontinued). Pfizer and Lilly will also consider requests for the protocol, data dictionary, and statistical analysis plan. Data may be requested from Pfizer trials 24 months after study completion. The de-identified participant data will be made available to researchers whose proposals meet the research criteria and other conditions, and for which an exception does not apply, via a secure portal. To gain access, data requestors must enter into a data access agreement with Pfizer.