Figures & data
Figure 1 Flow diagram illustrating the structure of a typical bioinformatics project. Note that, since putative lipoproteins typically represent ca. 2% of a given proteome, projects can be scaled to match different sized assessments by choosing genomes of different sizes or by allocation of only a proportion of the sequences recovered by pattern searching.
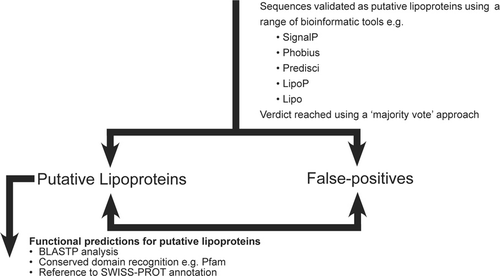
Figure 2 Sequence alignment illustating the problem of ‘putativism’. Sequence BSU_LSP is a 40 amino acid stretch of the Bacillus subtilis lipoprotein signal peptidase enzyme, covering the important conserved aspartate catalytic diad (CitationTjalsma et al., 1999). Theoretical sequence Q1 is 20/40 (50%) amino acids identical to BSU_LSP and might reasonably be annotated as a putative lipoprotein signal peptidase, since the crucial aspartates (▴) are conserved. Theoretical sequence Q2 is highly homologous to Q1 (20/40, 50% amino acid identity) and all three sequences are homologous to each other (* below alignment indicate amino acid identity). However, even though sequence Q2 is homologous to BSU_LSP (12/40, 30% amino acid identity) the critical aspartate catalytic diad is not conserved and it would be erroneous to annotate this sequence as a putative lipoprotein signal peptidase. This illustrates the flaw in the logic that because A is similar to B and B is similar to C, then A must have the same function as C: proteins A and C may have diverged to the point where they are functionally distinct. Allowing students to explore these concepts can be a valuable learning experience.

Table 1 Analysis of bioinformatic project marks compared to overall cohort mark over three academic years