Figures & data
Figure 1. Biochemical pathways of DNA methylation and demethylation. (A) Physiologic state. (I) DNMTs add a methyl group to cytosines of CpG-islands. (II) TET2 catalyzes the conversion of 5-mC to 5-hmC. (III) AID mediates degradation of 5-hmC to 5-hmU. (IV) 5-hmU activates the BER-pathway, in which TDG or SMUG1 enable further degradation to unmethylated cytosine. (V) TET2 can also convert 5-mC to 5-fC and 5-caC. (VI) Both 5-fC and 5-caC are directly recognized and repaired by TDG-mediated BER. (VII) IDH1/2 converts isocitrate to α-KG which is an essential cofactor for TET2-mediated conversion of 5-mC to 5-hmC. (B) Effect of commonly occurring mutations in enzymes involved in DNA methylation and demethylation. (I) Mutant TET2 is unable to convert 5-mC to 5-hmC, which results in decreased 5-hmC levels, and thus inhibits demethylation. (II) IDH1/2 gain-of-function mutations result in a neomorphic enzyme activity that converts α-KG to 2-HG, thus inhibiting α-KG-dependent functions of TET2. (III) 2-HG also inhibits the JMJC-family of HDMs, and thus inhibits demethylation of histones. Both TET2 loss-of-function and IDH1/2 gain-of-function mutations result in reduced 5-hmC levels and result in global promoter hypermethylation.
The green field demarks methylation, whereas the red field demarks occurring demethylation. The yellow lightning flashes denote molecules that are being evaluated as therapeutic targets in MDS/AML.
Abbreviations: 2-HG, 2-hydroxyglutarate; 5-caC, 5-carboxyl-cytosine; 5-fC, 5-formyl-cytosine; 5-hmC, 5-hydroxy-methyl-cytosine; 5-hmU, 5-hydroxy-methyl-uridine; 5-mC, 5-methyl-cytosine; AID, activation-induced cytidine deaminase; α-KG, α-ketoglutarate; AML, acute myeloid leukemia; BER, base excision repair pathway; DNMT, DNA methyltransferase; HDM, histone demethylase; IDH, isocitrate dehydrogenase; JMJC, jumonji-domain-containing; MDS, myelodysplastic syndrome; SAH, S-adenosylhomocysteine; SAM, S-adenosylmethionine; SMUG, single-strand selective monofunctional uracil DNA glycosilase; TDG, thymine DNA glycosilase; TET, ten-eleven translocation.
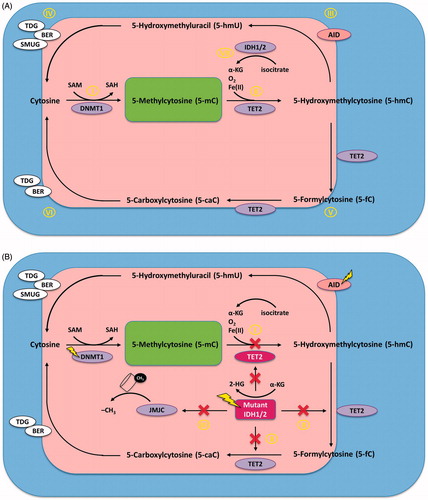
Figure 2. Enzyme network of epigenetic regulation of gene expression. The most relevant enzymatic systems known to modify DNA and/or histones are depicted. (A) DNMT3A and DNMT3B are responsible for de novo DNA methlyation. They catalize the addition of a methyl (CH3) group (denoted as “M”) at the 5-carbon atom of cytosine to form 5-mC in the context of CgP-islands in the promoter regions of genes. This blocks the access of transcription factors to the DNA and results in gene silencing. After binding methylated CpG, MeCP2 recruits and forms the MeCP2/DNMT3A-B/Sin3A/PU.1/HDAC1 co-repressor complex to silence transcription via histone deacetylation (denoted as “A”), which is mediated by HDAC1. JMJC is an HDM, which demethylates lysines 9 and 36 of histone H3. DNMT3L is catalytically inactive, but forms a complex with DNMT3A and recruits and enhances its activity to histone 3 when K4 is not methylated. (B) HMTs confer methylation of histone H3 at lysines 4, 36, and 79 (H3K4, H3K36 and H3K79) resulting in an open chromatin structure, which represents a permissive transcriptional state associated with gene activation. IDH2 enzymes hydroxylate isocitrate to α-KG, which is in turn utilized by TET2 to convert 5-mC to 5-hmC, ultimately resulting in DNA hypomethylation. HATs confer the acetylation of lysine residues on the N-terminus of histones, which is generally associated with active gene transcription. BRD proteins bind acetylated histones of target genes and activate transcription. (C) EZH2 is an H3K27 methyltransferase which requires formation of PRC2 to catalyze the addition of methyl groups to arginine and lysine residues on the N-terminal tail of histones. Methylation of histone H3 at lysines 9 and 27 (H3K9 and H3K27Me3) by the PRC2 complex represents a repressive transcriptional state associated with gene silencing. ASXL1 recruits and stabilizes the PRC2 complex at target locations in the genome. (D) HDMs demethylate H3K9 and H3K27 resulting in an open chromatin structure, and IDH2 and TET function along the same pathway to induce demethylaton of CgP-islands, ultimately resulting in a permissive transcriptional state. Most mutations of epigenetic modifiers in MDS and AML affect post-translational modifications on the N-terminal tail of histone 3 or at cytosines of DNA.
Purple enzymes are associated with loss-of-function mutations (DNMT3A, TET2, EZH2, ASXL1), whereas pink enzymes are associated with gain-of-function mutations of their respective genes (IDH1/2).
Abbreviations: 5-hmC, 5-hydroxy-methyl-cytosine; 5-mC, 5-methyl-cytosine; α-KG, α-ketoglutarate; AML, acute myeloid leukemia; ASXL1, additional sex combs like 1; BRD, bromodomain; DNMT, DNA methyltransferase; EZH2, enhancer of zeste homolog 2; HAT, histone acetyltransferase; HDAC1, histone deacatylase 1; HDM, histone demethylase; HMT, histone methyltransferase; IDH, isocitrate dehydrogenase; JMJC, jumonji-domain-containing; MDS, myelodysplastic syndrome; MeCP2, methyl CpG binding protein 2; PRC2, polycomb repressor binding complex 2; TET, ten-eleven translocation; TSG, tumor necrosis factor-stimulated genes.
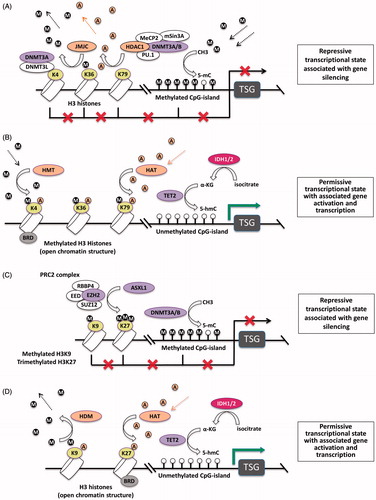
Table 1. Novel substances targeting the methylation machinery currently being evaluated in clinical trialsa.
Figure 3. Mechanisms of action of AZA and DAC. (A) DAC is incorporated into DNA as a substitute for cytosine. This results in covalent trapping and depletion of DNMT1 (and AID), as well as in loss of DNA methylation marks. This eventually results in DNA demethylation and (re-)activation of gene expression. Depending on which genes are re-induced, this may lead to: (i) induction of apoptosis, (ii) induction of differentiation, and/or (iii) induction of an effective host anti-leukemia immune response, all of which ultimately result in inhibition of malignant proliferation. (B) DNMT1 is localized in the nucleus when NLS and BAH are present. HMAs induce hyperphosphorylation of DNMT1 via PKCδ. Phosphorylated DNMT1 is then targeted to the ubiquitination machinery, a process which requires KEN-Box. This leads to proteasomal degradation of both DNMT1 and AID. Lower levels of AID result in elevated levels of AZA/DAC, which can then further reduce DNMT1 levels via all the mechanisms described in this figure. (C) About 80–90% of AZA is incorporated into RNA resulting in mRNA and protein metabolism disruption, both of which ultimately inhibit malignant proliferation.
The yellow lightning flashes denote molecules that are being evaluated as therapeutic targets in MDS/AML.
Abbreviations: AID, activation induced cytidine deaminase; AML, acute myeloid leukemia; AZA, 5-azacitidine; BAH, bromo-adjacent homology; DAC, 5-aza-2′-deoxycytidine; DNMT1, DNA methyltransferase 1; HDAC1, histone deacatylase 1; HMAs, hypomethylating agents; IFNα, interferon alpha; IL15, interleukin 15; MDS, myelodysplastic syndrome; MHC, major histocompatibility complex; NKCs, natural keller cells; NLS, nuclear localization signal; P, phosphate; PKCδ, protein kinase C delta; Tc1, Type 1 CD8+ T cells; Th1, Type 1 T helper cell; Th17, Type 17 T helper cell; Tregs, regulatory T cells; U, uridine; UHRF1, ubiquitin-like with PHD and ring finger domains 1.
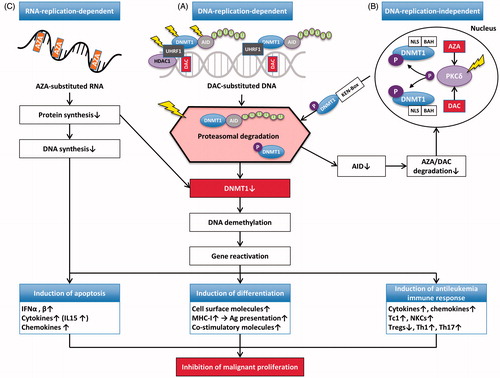
Table 2. DNA-replication-independent mechanisms of DNMT1 depletion.
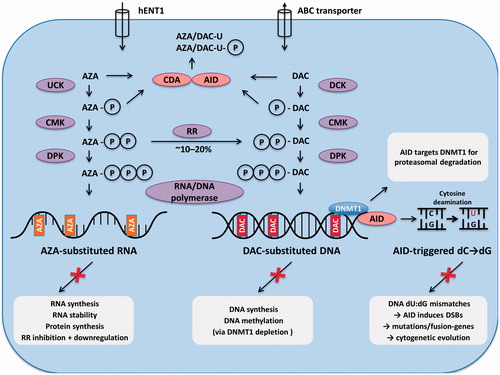
Figure 4. Membrane transporters and intracellular metabolism of AZA and DAC. AZA and DAC enter the cell via nucleoside transporters (e.g. hENT1). After triphosphorylation by the respective enzymes they are incorporated into RNA in the case of AZA, or into DNA in the case of DAC. Approximately 10–20% of AZA is reduced to DAC by RR, which is followed by incorporation into DNA. However, this step is self-limited and transient. Excess azanucleosides are rapidly deaminated to uracil-moieties by CDA. It is likely that AID can also perform this step. If the deamination process occurs on already DNA-incorporated DAC-cytidine residues, this will result in dU:dG mismatches on DNA, and may ultimately lead to DNA DSBs. AID-triggered DSBs can also be substrates for pro-oncogenic chromosomal translocations. AID may thus trigger leukemic evolution. In addition, AID targets DNMT1 for proteasomal degradation.
Abbreviations: ABC transporter, ATP-binding cassette transporter; AID, activation induced cytidine deaminase; AZA, 5-azacitidine; C, cytosine; CDA, cytidine deaminase; CMK, cytidine monophosphate kinase; DAC, 5-aza-2′-deoxycytidine; dC, deoxycytidine; DCK, deoxy-cytidine kinase; dG, deoxyguanine; DNMT1, DNA methyltransferase 1; DPK, diphosphate kinase; DSB, DNA double strand breaks; dU, deoxyuridine; G, guanine; hENT1, human equilibrative nucleoside transporter 1; P, phosphate; RR, ribonucleotide reductase; U, uridine; UCK, uridine-cytidine kinase.