Figures & data
Table I. Hereditary autoinflammatory diseases. (Modified after (Citation6)).
Figure 1. Signalling pathways activated by TNF-α binding to TNFR1. TNFR1 is expressed on the cell surface as a transmembrane receptor with four cysteine-rich domains (CRD) on the extracellular side and a death domain (DD) intracellularly. The first CRD is a pre-ligand assembly domain (PLAD) that interacts with PLADs from other TNFR1s to form receptor homotrimers, which without their ligand preferentially associate with inhibitory proteins in the cytoplasm, such as silencer of death domain (SODD) (Citation26,Citation28,Citation29). The binding of a TNF-α homotrimer leads to a change in relative positioning of the intracellular DDs, favouring assembly of a signalling complex by interaction of the TNFR1–TNF-α complex, TNFR1-associated death domain adaptor (TRADD) protein, receptor-interacting protein (RIP), and TNF-receptor-associated factor 2 (TRAF2) (Citation30). The interaction of this complex with other effector proteins leads to signalling through, among others, nicotinamide adenine dinucleotide phosphate oxidase (NADPH oxidase) and inhibitor of κB kinase complex (IKK complex), resulting in formation of reactive oxygen species (ROS) and liberation of nuclear factor kappa B (NF-κB) respectively, both of which contribute to the induction of the inflammatory response (Citation28,Citation31–34). The internalization of the signalling complex by endocytosis favours the assembly of a death-inducing signalling complex (DISC), consisting of the TNFR1–TNF-α complex, TRADD, Fas-associated death domain (FADD), and caspase-8, leading to the induction apoptosis, which in turn is inhibited by NF-κB activation (Citation30,Citation35). TNFR1 signalling is terminated by receptor endocytosis and degradation and post-translational modifications of the signal-transducing proteins (Citation36,Citation37). Cleavage of TNFR1 by matrix metalloproteinases (MMPs) on the cell surface results in the production of soluble TNFR1s (sTNFR1s) that are capable of binding free TNF-α trimers, possibly inhibiting TNF-mediated signalling (Citation37,Citation38).
This figure is partly based on the article by Kimberley et al. (Citation60).
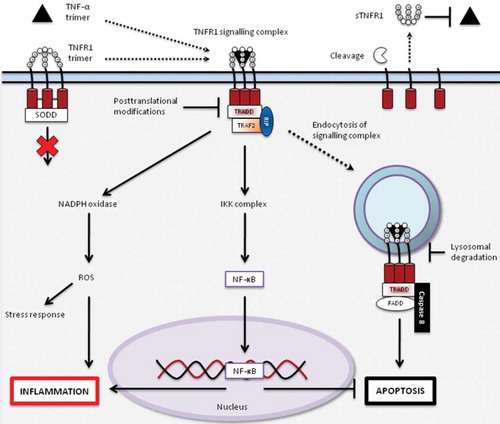
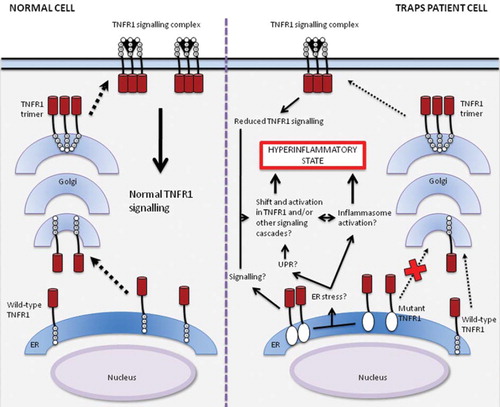
Figure 2. Model of signalling and trafficking pathways for wild-type and mutant TNFR1 in normal cells and cells from TRAPS patients. Wild-type receptors traffic from the endoplasmic reticulum (ER) through Golgi to the cell surface, allowing normal TNF-α-induced signalling. In cells from patients with TRAPS, mutant TNFR1s are retained in the ER (Citation39,Citation50,Citation51), where they may activate signalling pathways on their own or by inducing ER stress, leading to an unfolded protein response (UPR) (Citation52,Citation55,Citation56) or perhaps inflammasome activation (Citation23–25,Citation32), resulting in an inflammatory response. Trafficking and signalling of wild-type TNFR1s remains functional in cells of TRAPS patients, albeit reduced when compared to normal cells (Citation41,Citation42,Citation46). These mechanisms together could lead to a shift in balance of signalling and a self-perpetuating activation of TNFR1 or other signalling cascades by a positive feedback loop, producing the hyperinflammatory phenotype characteristic of TRAPS.
This figure is partly based on the article by Kimberley et al. (Citation60).