Figures & data
Figure 1. NO biosynthesis and vascular smooth muscle targets. In endothelial cells, the L-arginine–endothelial nitric oxide synthase (eNOS) system and the synthesis via the de-novo synthesis and salvage pathways of tetrahydrobiopterin (BH4), an essential co-factor of eNOS, are shown. ([Ca2+]i = intracellular calcium concentration; Ca2+-sensitivity = sensitivity to calcium of contractile proteins; cGMP = cyclic guanosine monophosphate; cGKI = cGMP-dependent protein kinase I; Cyt-C = cytochrome C; DHFR = dihydrofolate reductase; FAD = flavin adenine dinucleotide; FMN = flavin mononucleotide; GTP = guanosine triphosphate; NADPH = nicotinamide adenine dinucleotide phosphate; NO2- = nitrite ion; NO3- = nitrate ion; O2.- = superoxide anion; ONOO- = peroxynitrite; P450 = cytochrome P450 mono-oxygenases; SERCA = sarcoplasmic reticulum calcium ATPase; sGC = soluble guanylyl cyclase; XO = xanthine oxidase).
![Figure 1. NO biosynthesis and vascular smooth muscle targets. In endothelial cells, the L-arginine–endothelial nitric oxide synthase (eNOS) system and the synthesis via the de-novo synthesis and salvage pathways of tetrahydrobiopterin (BH4), an essential co-factor of eNOS, are shown. ([Ca2+]i = intracellular calcium concentration; Ca2+-sensitivity = sensitivity to calcium of contractile proteins; cGMP = cyclic guanosine monophosphate; cGKI = cGMP-dependent protein kinase I; Cyt-C = cytochrome C; DHFR = dihydrofolate reductase; FAD = flavin adenine dinucleotide; FMN = flavin mononucleotide; GTP = guanosine triphosphate; NADPH = nicotinamide adenine dinucleotide phosphate; NO2- = nitrite ion; NO3- = nitrate ion; O2.- = superoxide anion; ONOO- = peroxynitrite; P450 = cytochrome P450 mono-oxygenases; SERCA = sarcoplasmic reticulum calcium ATPase; sGC = soluble guanylyl cyclase; XO = xanthine oxidase).](/cms/asset/a16fd03e-aac8-4a72-9bb1-e8cd98a18391/iann_a_585658_f0001_b.gif)
Figure 2. L-arginine–NO-synthase–soluble guanylyl cyclase pathway and potential sites of therapeutic intervention. The circled numbers (from 1 to 14) indicate potential sites of therapeutic intervention. 1) L-arginine supplementation. 2) Inhibition of protein arginine N-methyltransferase type I (PRMT-I) to prevent the formation of asymmetric dimethyl-L-arginine (ADMA). 3) Increasing the expression and/or the activity of dimethylarginine dimethylaminohydrolase-2 (DDAH-2) to facilitate ADMA catabolism. 4) Inhibition of arginase-2 to prevent L-arginine metabolism. 5) Increasing the expression and/or activity of endothelial nitric oxide synthase (eNOS). 6) Designing drugs that stimulate endothelium-derived nitric oxide release. 7) Enhancing the expression and/or activity of guanosine triphosphate cyclohydrolase (GTPCH), the rate-limiting enzyme for tetrahydrobiopterin synthesis (BH4), or direct supplementation with BH4, or with its precursor sepiapterin. 8) Enhancing the expression and/or activity of dihydrofolate reductase (DHFR), involved in BH4 regeneration. 9) Scavengers of reactive oxygen species (ROS), anti-oxidants. 10) Inhibition of the activity and/or expression of enzymes that generate ROS such as NAD(P)H oxidases (NOX), cyclo-oxygenases (COX), lipoxygenases (LOX), or cytochrome P450 mono-oxygenases (P450). 11) Enhancing the expression and/or activity of enzymes that metabolized ROS such as superoxide dismutase (SOD) or catalase (or alternatively synthesis of mimetics). 12) Stimulators of soluble guanylyl cyclase (sGC). 13) Activators of sGC. 14) Inhibitors of phosphodiesterase-5 (PDE-5). (BH2 = dihydrobiopterin; CAT-1 = cationic amino acid transporters; CaV = voltage-activated calcium channel; cGMP = cyclic guanosine monophosphate; EC = endothelial cell; FAD = flavin adenine dinucleotide; FMN = flavin mononucleotide; O2.- = superoxide anion; ONOO- = peroxynitrite; PKG = protein kinase G; VSMC = vascular smooth muscle cell).
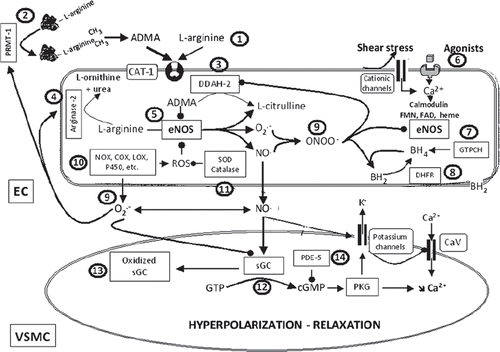
Figure 3. NO and EDHF. Endothelium-dependent relaxations resistant to inhibitors of NO synthases and cyclo-oxygenases, leading to the hyperpolarization of the underlying smooth muscle cells, are associated with different mechanisms: the metabolism of arachidonic acid via the lipoxygenases (LOX) or cytochrome P450 mono-oxygenases (P450) pathways and the non-chemical EDHF pathways, which, following the calcium-calmodulin-dependent activation of endothelial potassium channels, involve either the direct transfer of negative charges (e−) via the myoendothelial gap junction or the accumulation of potassium ions in the intercellular space and the subsequent activation of the smooth muscle Na+/K+-ATPase and/or the inwardly rectifying potassium channel. The circled numbers (from 1 to 7) indicate the principal steps in the development of endothelium-dependent hyperpolarizations and the potential sites of therapeutic intervention. The privileged therapeutic options, inhibitors of soluble epoxide hydrolase (sEH) and activators of endothelial potassium channels, are indicated by a broad arrow. 1) Agonists interacting with their receptor and physical forces such as shear stress increase endothelial intracellular calcium concentration. 2) Activation of transient receptor potential channels (TRP) contributes to calcium entry. 3) Activation of endothelial calcium-activated potassium channels of small (SKCa or KCa2.3) and intermediate conductance (IKCa or KCa3.1); the endothelial cell hyperpolarization increases the driving force for calcium entry. 4) Calcium-dependent activation of the endothelial nitric oxide synthase (eNOS); epoxyeicosatrienoic acids, P450 metabolites of arachidonic acid (AA), enhance NO production. 5) EETs and LOX metabolites, such as hydroxyeicosatetraenoic acids (HETE) and trihydroxyeicosatrienoic acids (THETA), can be released by endothelial cells and activate potassium channels of the smooth muscle cells to produce their hyperpolarization and relaxation. EETs are metabolized by sEH to form the generally less active dihydroxyeicosatrienoic acids (DHET). 6) Electrotonical coupling between endothelial and vascular smooth muscle cells via myoendothelial gap junctions clustered at specific endothelial projections toward the vascular smooth muscle cells. 7) Vascular smooth muscle hyperpolarization inhibits the activation of voltage-gated calcium channels (CaV) and decreases the intracellular calcium concentration.
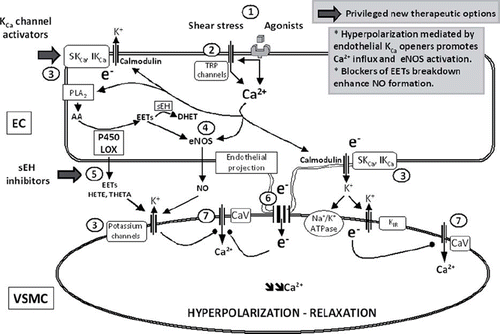
Figure 4. NO and endothelin. Release and actions of endothelin-1 (ET-1) in the vascular wall. (AA = arachidonic acid; AVP = arginine vasopressin; cAMP = cyclic AMP; cGMP = cyclic GMP; COX = cyclo-oxygenases; ECE = endothelin-converting enzyme; ETA and ETB = endothelin-receptors; NO = nitric oxide; NOS = nitric oxide synthase; PGI2 = prostacyclin; R = cell membrane receptor) (from Feletou et al., 2008, with permission). (Reference 9)
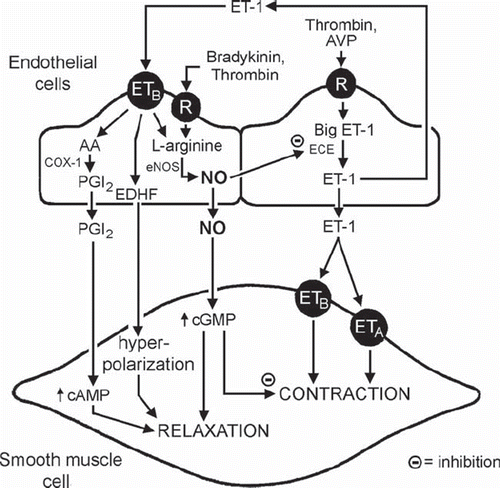
Figure 5. Endothelium-derived contracting factors (EDCF). Endothelium-dependent contraction is likely to be comprised of two components: generation of prostanoids and ROS. Each component depends on the activity of endothelial COX-1 and the stimulation of the TP receptors located on the smooth muscle to evoke contraction. In the SHR aorta, there is an increased expression of COX-1 and EP3 receptors, increased release of calcium, ROS, endoperoxides, and other prostanoids, which facilitates the greater occurrence of endothelium-dependent contraction in the hypertensive rat. The necessary increase in intracellular calcium can be triggered by receptor-dependent agonists, such as acetylcholine or ADP, or mimicked with calcium-increasing agents, such as the calcium ionophore A23187. The abnormal increase in intracellular ROS can be mimicked by the exogenous addition of H2O2 or the generation of extracellular ROS by incubation of xanthine with xanthine oxidase. The circled numbers indicate potential sites of therapeutic intervention: 1) inhibition of cyclo-oxygenases; and 2) antagonists of TP receptors. (AA = arachidonic acid; ACh = acetylcholine; ADP = adenosine diphosphate; H2O2 = hydrogen peroxide; m = muscarinic receptors; P2Y = purinergic receptor isoform 2Y; PGS, prostaglandin synthases; PGD2 = prostaglandin D2; PGE2 = prostaglandin E2; EP3, prostaglandin E2 receptor subtype 3; PGF2α = prostaglandin F2α; PGI2 = prostacyclin; PGIS = prostacyclin synthase; PLA2 = phospholipase A2; ROS = reactive oxygen species; TXA2 = thromboxane A2; TXAS = thromboxane synthase; X + XO = xanthine plus xanthine oxidase) (from Tang and Vanhoutte, 2009, with permission). (Reference 239)
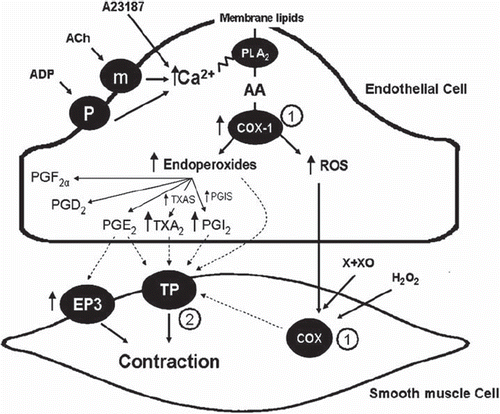
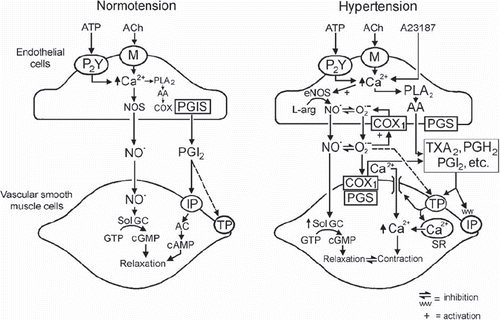
Figure 6. Endothelium-dependent effects of acetylcholine in rat aorta. Left: endothelium-dependent relaxations in normotensive rats. Right: cyclo-oxygenase-dependent, endothelium-dependent contractions to acetylcholine in SHR aorta. (AA = arachidonic acid; AC = adenylyl cyclase; COX1 = cyclo-oxygenase 1; IP = PGI2 receptor; M = muscarinic receptor; PGH2 = endoperoxides; PGI2 = prostacyclin; PGIS = prostacyclin synthase; PLA2 = phospholipase A2; R = receptor; sGC = soluble guanylyl cyclase; SR = sarcoplasmic reticulum; TP = TP receptor; + = activation; - = inhibition) (from Félétou and Vanhoutte 2006, with permission from the American Physiological Society). (Reference 1)