
Figures & data
Figure 1. Selection of patients for the treatment response analysis. PAH, pulmonary arterial hypertension; IPAH, idiopathic pulmonary arterial hypertension; HPAH, hereditary pulmonary arterial hypertension; CTD-PAH, connective tissue disease-associated pulmonary arterial hypertension; RHC, right heart catheterization.
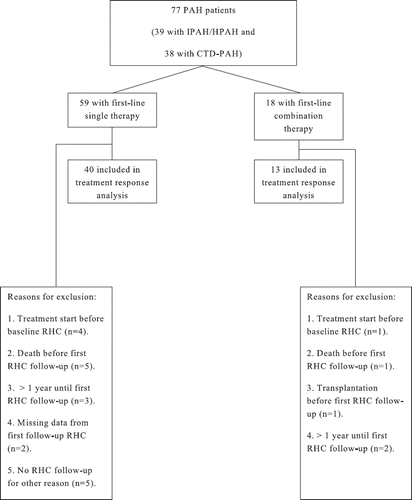
Table I. Characteristics at first right-heart catheterization for the entire study population.
Figure 2. 1-, 2- and 3-year survival rates, respectively, for (A) the entire study population, (B) IPAH/HPAH- and CTD-PAH patients, respectively, and the proportion of patients that started first-line single therapy and who were still alive on single therapy at 1-, 2- and 3-years, respectively, from treatment start for (C) the entire study population and (D) IPAH/HPAH- and CTD-PAH patients, respectively. IPAH, idiopathic pulmonary arterial hypertension; HPAH, hereditary pulmonary arterial hypertension; CTD-PAH, connective tissue disease-associated pulmonary arterial hypertension. Our findings support that survival has improved, as compared with untreated patients in the 1991 NIH registry study, which reported a median survival of 2.8 years for patients with primary pulmonary hypertension, and only approximately one year for patients with associated Raynaud phenomenon (A, B). Our data also show that a large proportion of patients who started on first-line single therapy required early treatment escalation (C, D).
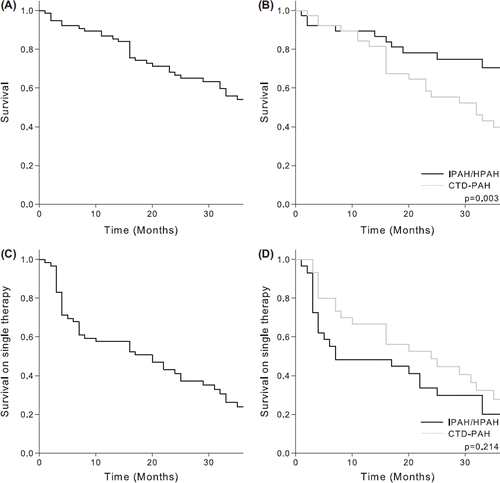
Table II. Baseline characteristics and haemodynamic response to first-line single- and combination therapy, respectively, in IPAH/HPAH and CTD-PAH.
Table III. Age-adjusted HR/SD for death or transplantation for variables at baseline and first RHC follow-up as well as of changes in variables from baseline to follow-up.