Figures & data
Figure 1. Primary and Secondary structure analyses of HP-1 light(α) and heavy(β) chains (HP1a and HP1b, respectively). The multiple sequence alignment show the identical (*), conserved (:) or semi conserved (.) aminoacid residues in the proteins analyzed by using the Clustal–Omega program (http://www.ebi.ac.uk/clustalo). HP was compared to similar proteins including the complement system proteases C1r (zymogen – 1gpza, and catalytic active domain – 1md8a) and C1s (1elva), apolipoprotein-H (1c1za), chymotrypsin (1ca0), elastase (1brup), chymase (1pjp), trypsin (1sgt), thrombin (1a2c) and coagulation factor X (1c5 m). The secondary structures were predicted by using the Jpred program (http://www.compbio.dundee.ac.uk/~www–jpred), and are shown in the protein sequence (β-strands in green, α-helices in red and extended regions in blue). Dashes represent gaps and the sequence C-terminal of complement system proteases C1r zymogen (1gpza) and C1s (1elva) and apolipoprotein-H (1c1za) was suppressed and represented by a gray block in HP1a sequence alignment. The binding sites for HB and the macrophage scavenger receptor CD163 (CD163) are marked in light pink and underlined, respectively. VPEKKT motif of the HP loop involved in the recognition of CD163 is marked in yellow. The position of the catalytic triad of the serine proteases are boxed and numbered according to the Trypsin superfamily. See the colored picture on the online version.

Figure 2. Comparison of the theoretical models of HP-1 light(α) and heavy(β) chains (HP1a and HP1b, respectively) with homologous proteins. Each protein secondary structures (β-strands in green, α-helices in red and extended regions in blue) on the left, and the electrostatic potential map (positive and negative regions in blue and red respectively) on the right were predicted by using Deepview/Swiss-PDB Viewer. See the colored picture on the online version.
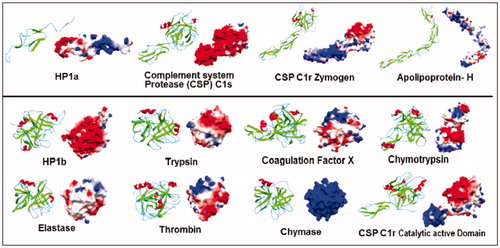
Figure 3. Comparison of the theoretical models of HP-1 monomeric (left) and dimeric (right) forms colored by the type of chain – light(α) and heavy(β) (yellow and purple, respectively), secondary structures (β-strands in green, α-helices in red and extended regions in blue) and electrostatic potential distribution (positive and negative regions in blue and red, respectively). Important structural regions are pointed including HB and macrophages CD163 receptor binding sites and the main disulfide bonds (arrows). See the colored picture on the online version.
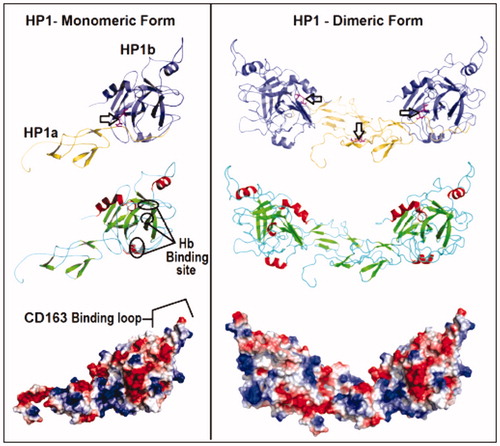
Figure 4. Structural analysis of the dimeric HP and ecotin-RR theoretical complex. Only one HP1 monomer (HP1a and HP1b) is depicted for clarity. HP1a is in yellow and HP1b in purple whereas ecotin β-strands are shown in light blue, α-helices in red and extended regions in light gray. The region of the interaction of ecotin-RR secondary binding site and HP1 residues are zoomed and show the hydrogen bonds (light green traces) formed on the right. Inset: Native PAGE of HP and ecotin-RR incubation leading to the complex H-R formation (up) and the Western-blotting assay using HP and Trypsin antibodies to recognize HP1 alone (down), reinforcing that HP is a Trypsin-related protein probably at both 3D-folding structure and immunogenic levels. See the colored picture on the online version.
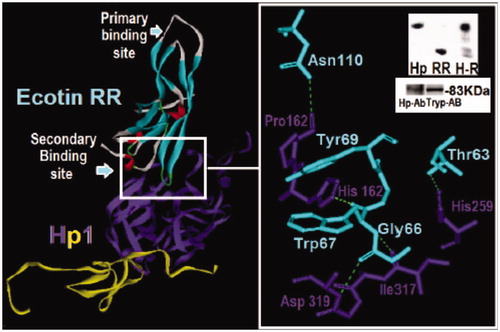
Figure 5. Comparison of ternary models formed by HP-1 dimer form (HP1) (blue), urokinase-type plasminogen activator (u-PA) (red) and wild-type ecotin (green) in (a) or with mutation in the primary binding site (M84R/M85R) (RR – dark yellow) in (b) or in both ecotin binding sites (D70R, V81T, T83S, M84R and M85R) (TSRR/R gray) in (c). The ecotin–aminoacid residues involved in HP and u-PA binding are in CPK. The insets report the number of aminoacid residues involved in hydrogen bonds that are higher for HP1 when a secondary binding mutation is added as in TSRR/R. The number (2) points the presence of two interactions simultaneously. See the colored picture on the online version.
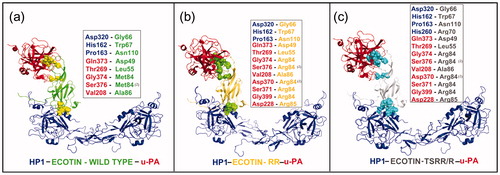
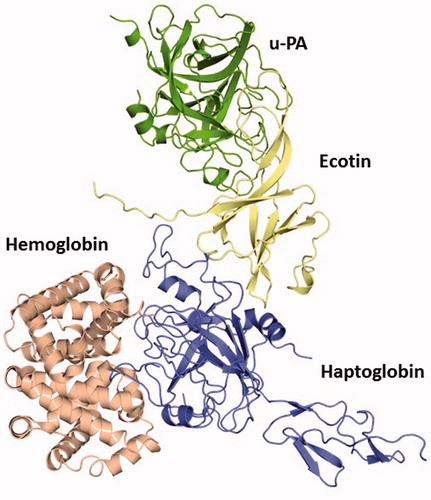
Figure 6. The quaternary complex HB–human HP–ecotin--uPA constructed based on the structural alignment with the crystal structure of the complex porcine HP–HB (PDB ID: 4F4O). This theoretical data revealed that even bound to ecotin, HP is able to establish feasible interactions to protect key HB residues from oxidative modification after exposure to hem-induced ROS. Thus, this complex points to the maintenance of HP physiological role despite its binding to this inhibitor. HB (light red), HP (blue), ecotin (yellow) and u-PA (green). See the colored picture on the online version.