Figures & data
Table I. ALS ideal biomarker characteristics.
Table II. Categories of biomarker in ALS.
Figure 1. Roadmap for biomarker discovery, biobanking and implementation of neurochemical markers in ALS.
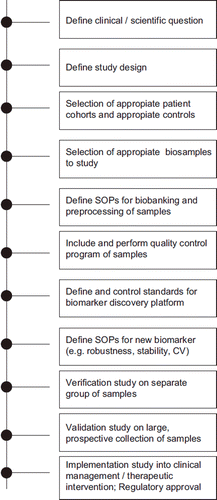
Table III. Types of control sample in ALS biomarker studies.
Table IV. Desirable minimum clinical dataset for ALS biomarker studies.
Table V. Strengths and weaknesses among biomarker sources.
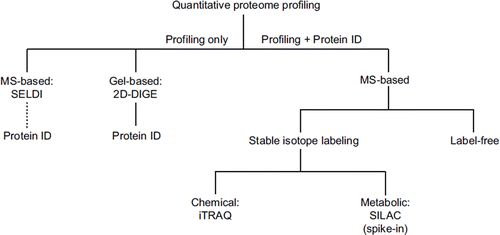
Figure 2. ‘Quantification tree’ showing the most common approaches for proteome profiling in discovery proteomics. SELDI and 2D-DIGE reveal only pattern information in the first instance, but 2D-DIGE (in contrast to SELDI) can be readily interfaced to mass spectrometric protein identification. MS-based approaches, which reveal both protein profile and identity, branch into stable isotope labeling-based and label-free techniques. Stable isotopes can be introduced by chemical tagging (e.g. iTRAQ) or by metabolic labeling (e.g. SILAC). SILAC can only be applied as a spike-in version to human material as it requires metabolic labeling of proteins in cell culture. For label-free quantification, peptide intensities are compared within separate LC-MS/MS runs.
SELDI: surface-enhanced laser desorption isonization; 2D-DIGE: two-dimensional fluorescence difference gel electrophoresis; MS: mass spectrometry; SILAC: stable isotope labeling with amino acids in cell culture; iTRAQ: isobaric tag for relative and absolute quantitation; LC: liquid chromatography.