Figures & data
Figure 1. Mammalian ISWI family chromatin remodeling complexes. (A) Depicted are the 7 currently known mammalian ISWI family ATP-dependent chromatin remodeling complexes. WICH, NoRC, RSF, ACF1 and CHRAC all share the SMARCA5 ATPase subunit. NURF and CERF share the SMARCA1 ATPase subunit. (B) Schematic representation of the domains within SMARCA1/SMARCA5. The catalytic activity of both SMARCA1 and SMARCA5 is defined by its ATPase domain that is split in 2 parts: DExx and HELICc. This domain, which is located in the N-terminal half, is part of the superfamily of DEAD⁄H-helicases and is shared by SWI2/SNF2-like ATP dependent chromatin remodelers. In addition, both proteins contain HAND, SANT and SLIDE domains that are characteristic for the ISWI family in their C-terminus. These domains regulate the activity of the ATPase domain and are involved in DNA binding.
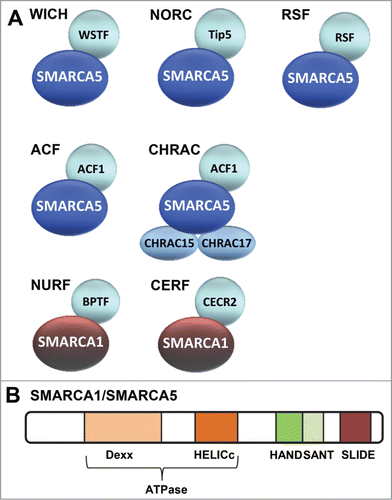
Figure 2. Mechanisms of double strand break and nucleotide excision repair. (A) Most double strand breaks (DSBs) are repaired either via Non-Homologous End-Joining (NHEJ) or Homologous Recombination (HR) and their detection initiates a DNA damage signaling cascade mediated by the PI3 kinase ATM. In NHEJ, detection of the break by Ku70/80 leads to recruitment of downstream factors including MRN, Artemis, and DNA-PK that process the DNA ends that are finally joined by XLF/XRCC4/LigIV. In HR, detection of the break by MRN leads to 5'-3' end resection, assisted by CtIP and EXO1, which creates ssDNA coated by RPA. RPA is replaced by RAD51, which mediates strand invasion and pairing of a homologous DNA strand that serves as template for error-free repair. The invading DNA end is extended by novel DNA synthesis and either dissociates and re-anneals with its original template, or a second DNA end invades the homologous template after which the resultant joint structures are resolved by specific endonucleases. Finally ligation takes place to complete repair. (B) UV-induced photolesions, monoadducts and other bulky lesions that distort the double helix are repaired by Nucleotide Excision Repair (NER). Lesions are either detected by the global genome NER machinery, via the concerted action of the UV-DDB and XPC/RAD23B complexes, or by the transcription-coupled NER machinery, involving CSA, CSB and UVSSA/USP7 that are recruited to DNA-damage-stalled RNA Polymerase II (RNApolII). Lesion detection leads to recruitment of the transcription factor H (TFIIH) complex, which unwinds DNA and verifies the presence of DNA damage. XPA binds to the lesion in the unwound DNA and RPA covers the non-damaged strand. The endonucleases ERCC1/XPF and XPG bind at both sites of the lesion and excise the damaged strand. The resulting gap is filled in by DNA synthesis and sealed by ligases.
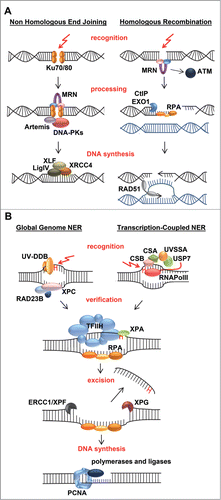
Figure 3. ISWI chromatin remodeling complexes in double strand break repair. Depicted is a model that includes the various functions of different ISWI complexes at sites of double strand break (DSB) repair. DSBs activate the PI3 kinase ATM, which phosphorylates histone variant H2AX at S139 (indicated with S-P), leading to the recruitment of MDC1, RNF8 and RNF168, which mediate a ubiquitylation signaling cascade. RNF168 is PARylated by PARP1 and interacts with SMARCA5. SMARCA5, whose recruitment is also regulated by NuMA and PARP3, stimulates RNF168-mediated histone ubiquitylation (indicated with Ub). At DSBs, SIRT6 deacetylates H3 at K56 (indicated with Ac), after which RNF20 is recruited to ubiquitylate H2B at K120 (indicated with Ub), coinciding with the methylation of H3 at K4 (indicated with M) and recruitment of SMARCA5, which interacts with SIRT6. SMARCA5 is furthermore recruited together with ACF1, CHRAC15 and CHRAC17 as part of the CHRAC complex and with WSTF as part of the WICH complex. WSTF interacts with and phosphorylates H2AX at Y142 (indicated with Y-P) to maintain S139 phosphorylation (indicated with S-P). Finally, RSF promotes the loading of histone-fold proteins CENP-S and CENP-X at or near DSB sites independently of SMARCA5. The recruitment of SMARCA5, ACF1, WSTF and RSF likely leads to the remodeling of chromatin that is necessary for efficient recruitment of repair factors, including BRCA1, RAD51, Ku70/80 and XRCC4, to facilitate repair by homologous recombination and non-homologous end-joining.
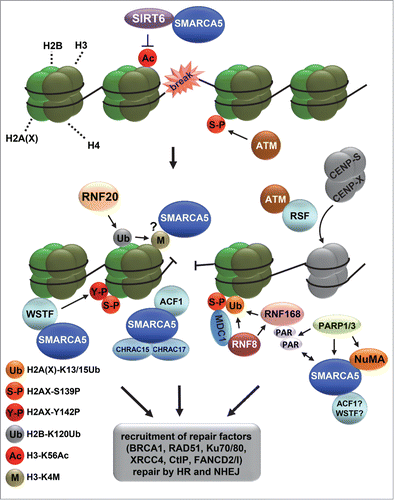
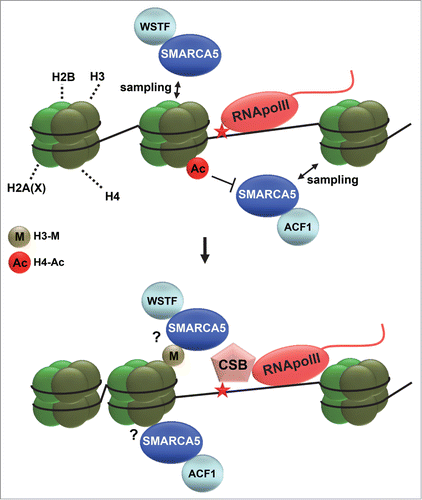
Figure 4. ISWI chromatin remodeling during NER. Depicted is a hypothetical model of how ISWI complexes function in transcription-coupled nucleotide excision repair. ISWI complexes, including ACF (SMARCA5 and ACF1) and WICH (SMARCA5 and WSTF), continuously sample DNA and only associate with chromatin when encountering specific signals. Such signals could be dependent on RNA polymerase II (RNApolII) arrest at DNA damage and could involve histone deacetylation (acetylation is indicated with Ac) and methylation (indicated with M). DNA damage arrested RNApolII binds with more affinity to the essential repair protein CSB and is itself reverse translocated, likely to make the lesion accessible for repair. SMARCA5 is necessary for efficient loading of CSB at sites of UV-induced transcriptional arrest, suggesting that chromatin remodeling facilitates access to DNA. Reverse translocation of RNApolII probably also requires chromatin remodeling.