Figures & data
Figure 1. Comparison of sequencing-based methods for genome-wide methylation analysis. (A) Overview of major next-generation sequencing-based methods for DNA methylation screening. Due to their practical relevance, we have also included quantitative array-based methylation screening methods. (B) General workflow for the most relevant bisulfite sequencing based methods RRBS, classic WGBS, PBAT and TWGBS. For methodological detail, please refer to the original reports describing the respective methods.Citation40-42,51,55-57 (C) Table showing advantages and disadvantages for bisulfite sequencing methods.
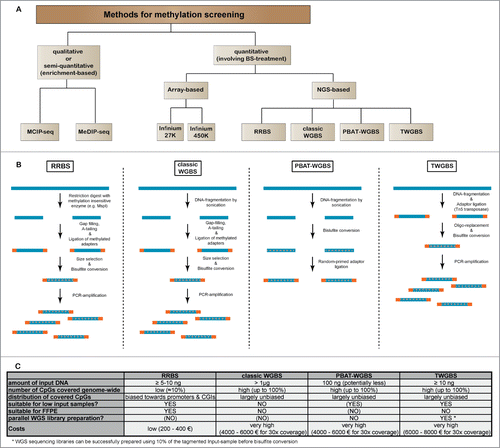
Figure 2. For figure legend, see page 3481.Figure 2 (See previous page). Conserved haematopoietic tissue-specific DNA-methylation. (A) Heatmap of all DMRs (n = 15,987) detected between the 4 HSPC populations (HSC, MPP1, MPP2 and MPP3/4). Each horizontal dash represents the ratio of methylated/unmethylated CpGs in each cell population. R1-R3: biological replicates. Nine distinct DNA methylation clusters were identified by unsupervised cluster analysis and can be grouped into 3 categories: Clusters 1–4 demonstrate a loss of methylation (blue) and clusters 6–9 a gain of methylation (red) from HSC to MPP3/4. Cluster 5 shows a loss of methylation from HSC to MPP2 followed by a gain of methylation to MPP3/4 (purple). (B) Display of mean methylation differences (ΔMeth) ±SD in DMRs of each cluster. ΔMeth is defined as the mean difference in methylation levels in a given subpopulation relative to the mean methylation level of all 4 subpopulations. Blue arrow downwards: loss of methylation; red arrow upwards: gain of methylation; purple arrows: bidirectional methylation change. N indicates the number of DMRs in each cluster. (C) Gene-ontology (GO) analysis of DMRs from clusters 1–9 (seeA) using GREAT with default association rule settings and “whole genome” as background.Citation69 Displayed are the enrichments of the 5 most significantly overrepresented GO-terms (P-value) for each cluster. Colors represent standard scores of log2 P-values. Rows were normalized to have a mean of 0 and a standard deviation of 1. (D) Analysis of DNA methylation changes across different tissues. Relative DNA methylation levels for all DMRs detected during early haematopoietic differentiation are displayed as a heatmap for 24 different cell and tissue types. DNA methylation levels were normalized relative to the mean level of all DMRs within each sample (blue and red indicate DNA methylation lower and higher than the mean methylation level, respectively). Samples analyzed include HSC, MPP1, MPP2 and MPP3/4 (3 replicates each; (R1-R3)), mouse embryonic stem cell data from 3 different experiments (mESC_XT, mESC_E14, and mESC_E14r) and WGBS data for 17 murine adult tissues.Citation66,78 DMRs are sorted as in A according to clusters 1–9.
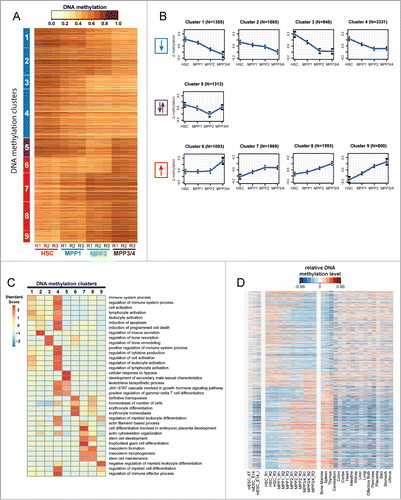
Figure 3. DNA-methylation changes correlate with gene-expression. (A) Left panel: Gene set enrichment analysis (GSEA) of genes associated with DMRs using the GSEA tool (Subramanian et al., 2005). GSEA is provided for DMRs identified between HSC−MPP1, MPP1−MPP2, MPP1−MPP3/4 and split into gain of methylation (red) or loss of methylation (blue) DMRs. NES: normalized enrichment score, FDR: false discovery rate. Right panel: Relative gene expression levels of core enriched set of genes normalized per row (purple: upregulated; green: downregulated). (B) Protein-protein interaction network based on GeneMANIA depicting the interactions of the “core enriched genes” for the HSC−MPP1 transition correlating with gain of methylation and decreased expression. The interaction network of the “core enriched genes” for the HSC−MPP1 transition correlating with loss of methylation and increased expression is depicted in Figure S1.
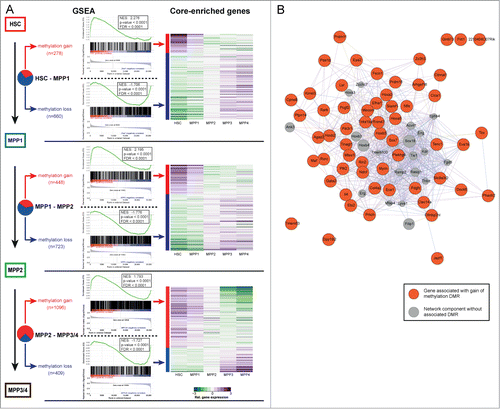
Figure 4. Correlation of DNA methylation and expression of transcription factors. (A) DMR frequency in 10 kb windows around the transcription start site (TSS) of transcription factors (TF) either differentially expressed (brown) or not differentially expressed (beige). The bottom part displays a heatmap of DMRs around TSS of TFs sorted according to P-value for overall differential expression within the HSPC compartment. Red dashes represent DMRs. Asterisks indicate statistical significance (Fisher's exact test; *: P-value 0.01–0.001; **: P-value <0 .001). (B) Left panel: Number of differentially expressed TFs in the transitions from HSC−MPP1, MPP1−MPP2 and MPP1−MPP3/4. Differentially expressed TFs with a DMR annotated are depicted in red, TFs without DMR are depicted in gray. Statistical enrichment of DMRs in differentially expressed TFs was calculated using Chi-squared test. Right panel: Mean expression values based on RNA-seq dataCitation5 of 4 representative differentially expressed TFs associated with DMRs are shown for each commitment step.
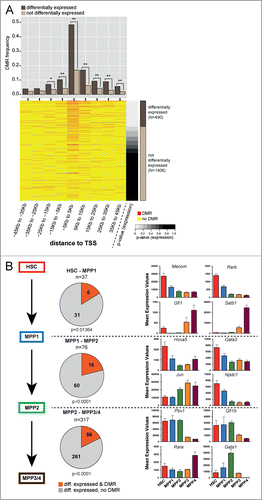
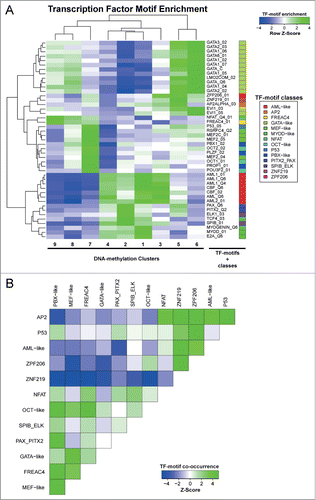
Figure 5. Motif enrichment in differentially methylated regions associated with transcription factors. (A) Cluster analysis of row Z-scores for TF motif enrichments in DNA methylation clusters. All DMRs were scored using 981 TRANSFAC position weight matrices and motifs showing a significant enrichment (Z > 2) or depletion (Z < −2) in any of the DMR clusters are displayed. Green: motif enrichment, blue: motif depletion. (B) Cluster analysis of Z-scores for pairwise TF-motif co-occurrences in all DMRs (see Material and Methods for details). Green: enrichment of TF-motif co-occurrence, blue: depletion of TF-motif co-occurrence.