Figures & data
Figure 1. A dynamic equilibrium between inducible CDKs and PP2A modulate the phosphorylation state of pocket proteins through the cell cycle. (A) Pocket proteins are active in their hypophosphorylated state. Pocket proteins are hypophosphorylated in early to mid G1 or in G0, when they are found associated with E2F/DP complexes and other proteins. Hypophosphorylated pocket proteins are also thought to bind transcription factors involved in differentiation (not shown). Pocket proteins are inactivated by CDK-dependent hyperphosphorylation. G1 Cyclin D1/CDKs start the process, which is coordinated with G1/S Cyclin E/CDK2 and maintained through S phase and mitosis by Cyclin A/CDK2 and Cyclin B/CDK1. PP2A and PP1 oppose the effects of CDKs. PP2A is active toward the 3 pocket proteins through the cell cycle and in quiescent cells, and this activity may be mediated by the cooperation of various trimeric PP2A holoenzymes (B(s)/PP2A) most prominently PP2A/B55α and PP2A/B55δ and perhaps PP2A/PR70 (see details in the text and 1B). This activity is regulated by a variety of signals and likely down-modulated in mitosis (see text for details). The hyperphosphorylation mediated by inducible CDKs, inactivates pocket proteins by disrupting/preventing their association with E2Fs, which mediates passage through the restriction point by triggering the expression of E2F-dependent (E2Fd) genes needed for DNA synthesis and, later, mitosis. When CDKs are inactivated late in mitosis (ML), B55/PP2A and in the case of pRB, PP1 abruptly dephosphorylate pocket proteins, resetting them to their active state (G1E, designates ealy G1). Arrows show the cell cycle activity span of the kinase or phosphatase with the same color. (B) B55α/PP2A appears to be one of the trimeric holoenzymes in equilibrium with CDKs through the cell cycle. This holoenzyme primarily targets p107 and p130 to a lesser extent. Since an interaction of this holoenzyme with pRB in chondrocytes has been detected, it is possible that it modulates all 3 pocket proteins, although likely with differential affinity. PR70/PP2A interacts with pRB in cells and with pRB and p130 in vitro. It remains to be determined if PR70/PP2A holoenzymes contribute to maintain the equilibrium with CDKs during the cell cycle or only target pRB (and perhaps the other pocket proteins) in response to certain stimuli (i.e., oxidative stress).
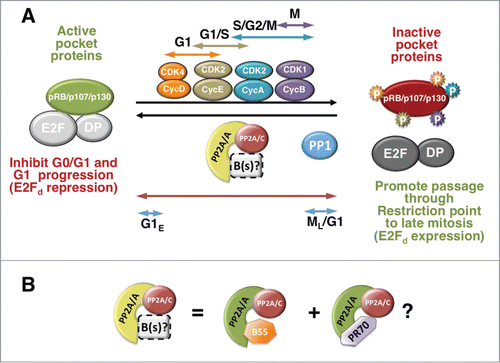
Figure 2. Trimeric PP2A holoenzyme and core dimer composition. The cartoon depicts the 4 types of trimeric holoenzymes and the core dimer. All the B regulatory subunit genes in each family are listed. The crystal structure of the core dimer and the trimeric holoenzymes containing B (B55α), B’ (B56γ) or B" (PR70) have been solved (see text for details). The bending of the scaffold changes with each B subunit, and the bending is maximum with PR70. It is thought that the particular B/C interfaces create B-family specific pockets for substrates. The structure of the B"’/PP2A holoenzymes has not ben solved (represented by a dashed B"’ subunit). B55α is a β-propeller with 7 blades and make less contacts with the catalytic subunit than B56γ. B56γ contains HEAT repeats like the scaffold. The PR70 structure is elongated and contains calcium binding sites, represented by green spheres. See text for additional details.
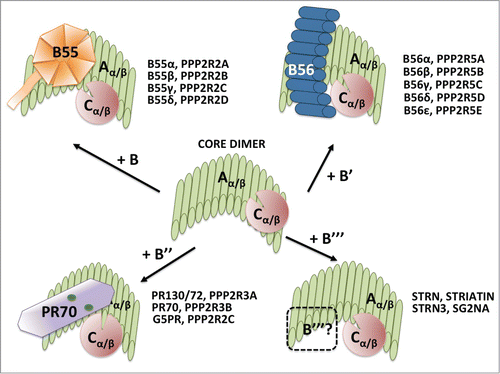
Table 1. PP2 A holoenzymes targeting pocket proteins during the cell cycle and in response to signals
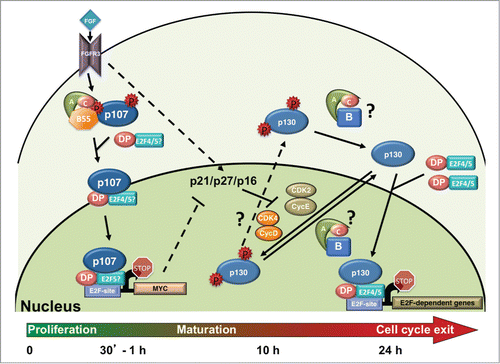
Figure 3. FGF induces rapid activation of B55α/PP2A holoenzymes, p107 dephosphorylation and activation, and cell cycle exit and maturation in chondrocytes. FGF stimulation through the FGFR3 receptor in chondrocytes leads to increased but transient formation of a B55α-δ/PP2A holoenzyme complexes with p107. This results in a shift on p107 localization from predominantly cytoplasmic to nuclear, formation of complexes with E2F4 and likely other E2Fs, and rapid recruitment to the c-MYC promoter, coinciding with its downregulation. p130 remains hyperphosphorylated as CDK4 and CDK2 remain active and may not be actively targeted by B55α/PP2A holoenzymes to switch the equilibrium toward dephosphorylation until CDK activity decreases. p21, p27 and p16 CKI activities increase by different means and appear to trigger inactivation of CDK4 and CDK2 coinciding with p130 and pRB dephosphorylation that occurs 10-15 hours post FGF stimulation. By 24 hrs. chondrocytes have exited the cell cycle and p130 and E2F4 are found at the promoters of cell cycle genes. Because hyperphosphorylated p107 is only detected in the cytoplasmic cellular fractions and this form is rapidly downregulated in the cytoplasm concomitantly with appearance of hypophosphorylated p107 in the nucleus upon FGF stimulation, it seems likely that this dephosphorylation occurs in the cytoplasm. Whether the same is true for p130 is less clear. p130 levels are very low in the absence of FGF stimulation and do not accumulate in the nucleus until several hours post-FGF treatment. While p130 phosphorylation by CDKs is likely occurring in the nucleus, the dephosphorylation step could conceivably occur either in the nucleus or the cytoplasm, hence the question marks for the PP2A reactions and the shuttling of phosphorylated p130 into the cytoplasm.Citation39 See text for additional details.