Figures & data
Figure 1. Flowchart of this investigation.
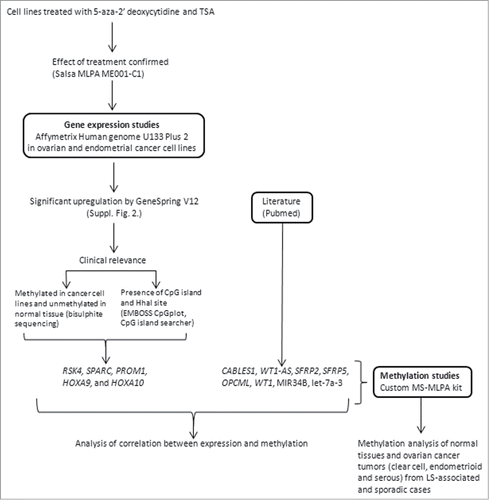
Figure 2. Average Dm values from MS-MLPA analyses on non-serous and serous ovarian carcinomas and the corresponding normal tissue references. Asterisks denote significantly elevated methylation in tumor vs. normal tissue by t-test for independent samples. The average Dm values of tumor DNAs may in fact be somewhat higher than those shown if possible “contamination” with normal cells is taken into account (see Materials and Methods).
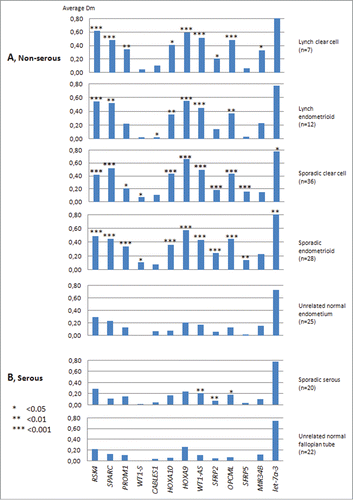
Table 1. Clinicopathological data of sporadic and Lynch-associated ovarian carcinoma
Table 2. Hypermethylation frequencies in different patient groups
Figure 3. Distribution of methylation dosage ratios (Dm values) for RSK4, SPARC, and HOXA9 in endometrioid ovarian carcinomas (sporadic and Lynch-associated combined) stratified by grade (low refers to grade 1 and high to grades 2 and 3). The horizontal line denotes the median and each triangle represents the Dm value of individual data point. Significance values by t-test for independent samples are shown.
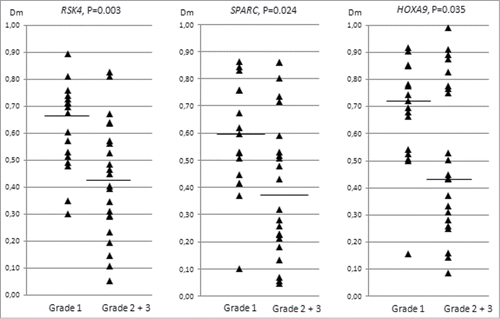
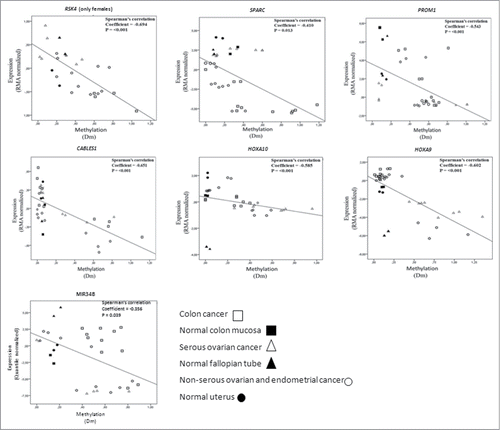
Figure 4. Correlation analysis of expression (Y-axis, RMA normalized values for protein coding genes and quantile normalized values for MIR34B from arrays) and methylation (X-axis, Dm values from MS-MLPA). The analysis includes cancer cell lines and normal tissue references for which high molecular weight DNA and RNA were available. Data points for normal tissues predominantly clustered in the left top quadrants, compatible with low methylation and high expression. Cancer cells with high degree of methylation often showed low expression (hence, were located in the right bottom quadrants), whereas cancer cells with low methylation showed high or low expression depending on the intrinsic properties of the genes and tissue types in question. While methylation for all these genes significantly correlated with transcriptional repression overall, subsets of specimens occasionally showed transcriptional regulation apparently unrelated to methylation (see, e.g., MIR34B).