Figures & data
Figure 1. IFNB1 induced autophagy in MCF-7 breast cancer cells by mediating MAP1LC3B conversion. (A) MCF-7 cells were IFNB1 responsive. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h, serum starved 3 h and stimulated with control medium or 100 or 1000 U/ml of IFNB1 for 30 min. Western blot analysis was performed for p-STAT1 and STAT1 protein. (B and C) IFNB1 induced MAP1LC3B-I to MAP1LC3B-II conversion. (B) MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and then treated with control medium, 1000 U/ml IFNB1 or 1 μM rapamycin for further 24 h. Western blot analysis was performed for MAP1LC3B and ACTB/β-actin proteins. (C) Quantification of band intensities in (B). Data represent mean and SEM of three independent experiments. Statistical analysis was performed using one-way repeated measures ANOVA followed by Dunett’s post-test against the control sample, *p < 0.05 and **p < 0.01. (D–F) IFNB1 induced eGFP-MAP1LC3B translocation. MCF-7 eGFP-MAP1LC3B cells were cultured for 72 h and then treated for an additional 24 h with control medium or 100 U/ml IFNB1 or with 200 nM rapamycin 2 h prior to fixation. Percentage of eGFP-MAP1LC3B-positive cells was quantified automatically as described in Materials and Methods. (D) Representative pictures of control, IFNB1 and rapamycin-treated cells. (E) Quantification of eGFP-MAP1LC3B translocation after IFNB1 treatment. Three different thresholds, >5, >10 or >15 eGFP-MAP1LC3B puncta/cell, were used to define an eGFP-MAP1LC3B puncta-positive cell. Data represent mean and SEM of three independent experiments, each obtained from an average of five replicates. Statistical analysis was performed using Student’s paired t-test, *p < 0.05. (F) Quantification of eGFP-MAP1LC3B translocation after rapamycin treatment. A threshold of >5 eGFP-MAP1LC3B puncta/cell was used to define an eGFP-MAP1LC3B puncta-positive cell. Data represent mean and SD of three replicates. Statistical analysis was performed using Student’s unpaired t-test, ***p < 0.001. (G) IFNB1 induced autophagic flow. MCF-7 RLuc-MAP1LC3B WT and RLucMAP1LC3BG120A cells were cultured for 72 h and then treated with control medium, 100 or 1000 U/ml IFNB1 or 200 nM rapamycin. The autophagic flux was measured at 6, 12 and 24 h after treatment as described in Materials and Methods. Data represent mean and SEM of six replicates and is representative of two independent experiments. Statistical analysis using one-way repeated measures ANOVA followed by Dunett’s post test against the control sample was performed individually for each time point, *p < 0.05, **p < 0.01 and ***p < 0.001.
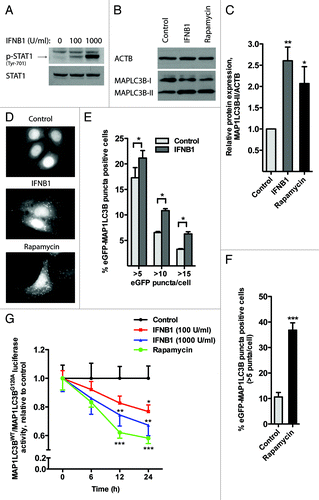
Figure 2. IFNB1 induced autophagy in MCF-7 breast cancer cells as measured by SQSTM1 degradation. (A–C) IFNB1 treatment triggered SQSTM1 degradation. (A) MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and then treated with control medium, 1000 U/ml IFNB1 or 1 μM rapamycin for 24 h. Western blot analysis was performed for SQSTM1 and VCL/vinculin protein levels. (B) Quantification of band intensities in (A). Data represent mean and SEM of five independent experiments. Statistical analysis was performed using one-way repeated measures ANOVA followed by Dunett’s post test against the control sample, ***p < 0.001. (C) MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and then treated with control medium or 1000 U/ml IFNB1 for the indicated time intervals. Western blot analysis was performed for SQSTM1 and ACTB levels. (D) IFNB1 did not regulate SQSTM1 mRNA levels. MCF-7 eGFP-MAP1LC3B cells were cultured and treated as in (A) before RNA was extracted and qPCR used to analyze SQSTM1 and ACTB levels. Data represent mean and SEM of two independent experiments.
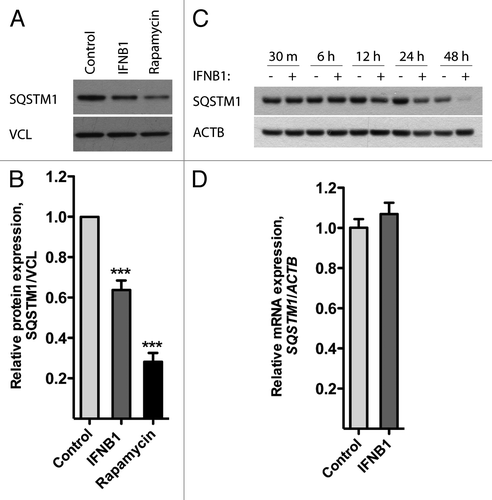
Figure 3. IFNB1 induced autophagy in MDAMB231 and SKBR3 breast cancer cells. (A) MDAMB231 cells were IFNB1-responsive. MDAMB231 cells were cultured for 24 h, serum starved for 3 h and stimulated with control medium or 1000 U/ml of IFNB1 for 30 min. Western blot analysis was performed for p-STAT1, STAT1 and ACTB proteins. (B and C) IFNB1 induced autophagy in MDAMB231 cells. (B) MDAMB231 cells were cultured for 24 h and then treated with control medium or 1000 U/ml IFNB1 for the indicated time intervals. Western blot analysis was performed for SQSTM1, MAP1LC3B and ACTB protein levels. The blot is representative of two independent experiments. (C) Quantification of the results shown in (B). The values are normalized to the time-point control, and further to the 30 min time-point set to 1, and represent average values from two independent experiments. The full line shows relative MAP1LC3B-II/ACTB levels, the dotted line shows relative MAP1LC3B-I/MAP1LC3B-II levels and the dashed line shows relative SQSTM1/ACTB levels. (D) SKBR3 cells were IFNB1-responsive. SKBR3 cells were cultured for 24 h, serum starved for 3 h and stimulated with control medium or 1000 U/ml of IFNB1 for 30 min. Western blot analysis was performed for p-STAT1, STAT1 and ACTB protein levels. (E and F) IFNB1 induces autophagy in SKBR3 cells. (E) SKBR3 cells were cultured for 24 h and then treated with control medium or 1000 U/ml IFNB1 for the indicated time intervals. Western blot analysis was performed for SQSTM1, MAP1LC3B and ACTB levels. The blot is representative of two independent experiments. (F) Quantification of the results in (E), as described in (C).
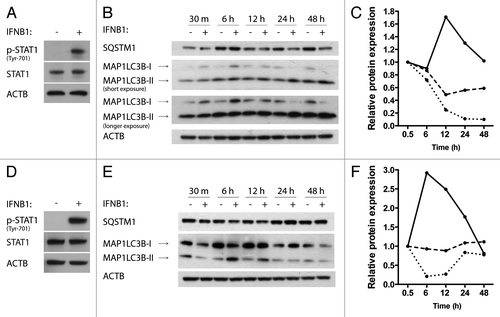
Figure 4. IFNB1 affected MTORC1 activity. (A and B) IFNB1 specifically decreased p-EIF4EBP1 levels. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and then treated with (A) control medium or 1000 U/ml IFNB1 for the indicated time intervals, or (B) with control medium or 100 or 1000 U/ml IFNB1 for 24 h. Western blot analysis was performed for p-EIF4EBP1, EIF4EBP1, p-RPS6, RPS6, ACTB and VCL protein levels. (C) Rapamycin triggered SQSTM1 degradation in a dose-dependent manner. Cells were cultured as in (A) and stimulated with the indicated concentrations of rapamycin for 24 h in order to find a saturating and a nonsaturating concentration of rapamycin. Western blot analysis was performed for SQSTM1 and ACTB protein levels. (D) IFNB1 did not potentiate rapamycin-mediated SQSTM1 degradation. Cells were cultured as in (A) and stimulated with control medium, 1000 U/ml IFNB1 and/or 1 μM rapamycin for 24 h. Western blot analysis was performed for SQSTM1 and ACTB protein levels. Shown is a representative western blot for SQSTM1 and ACTB protein levels of two independent experiments. Band intensities were quantified and the average relative intensities shown below. (E) IFNB1 and rapamycin acted together to degrade SQSTM1 at subsaturated concentrations. Cells were cultured as in (A) and stimulated with 100 U/ml IFNB1 and/or 0.1 nM rapamycin for 24 h. Western blot analysis was performed for SQSTM1 and ACTB protein levels. Band intensities were quantified and the relative quantifications are indicated.
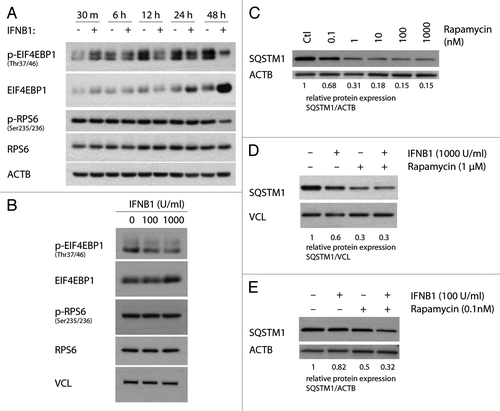
Figure 5. IFNB1 inhibited cell proliferation. (A) IFNB1 decreased cell numbers. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and then treated for 24 or 48 h with control medium or IFNB1 as indicated before a crystal violet assay was performed. Data represent mean and SEM of four replicates and is representative of two independent experiments. Statistical analysis was performed for each time point using one-way repeated measures ANOVA followed by Dunett’s post test against the control sample, ***p < 0.001. (B) IFNB1 inhibited proliferation. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and then treated for further 48 h with control medium or 1000 U/ml IFNB1. Cell proliferation was measured by means of BrdU incorporation. Data represent mean and SEM in arbitrary units (A.U.) of three independent experiments each obtained from an average of four replicates. Statistical analysis was performed using Student’s paired t-test, **p < 0.01. (C) IFNB1 induced G1-phase accumulation. MCF-7 eGFP-MAP1LC3B cells were cultured and treated as in (B). Then cells were stained with PI and cell cycle was analyzed by FACS. Red line is control treatment, blue line is IFNB1 treatment. The experiment is representative of two independent experiments.
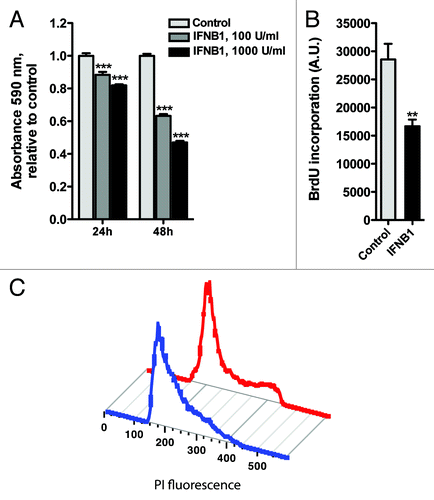
Figure 6. IFNB1 induced apoptosis. (A–E) IFNB1 induced apoptosis. (A) MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and then treated for further 48 h with control medium or 1000 U/ml IFNB1 and stained for DNA fragmentation using a TUNEL assay. TUNEL-positive cells were quantified as described in Materials and Methods. Data represent mean and SEM from three independent experiments. Statistical analysis was performed using Student’s paired t-test, **p < 0.01. (B and C) Representative western blots of cells cultured and treated as in (A) showing CASP8, PARP1, BAX and VCL protein levels. Blots are representative of three independent experiments. (D) MCF-7 eGFP-MAP1LC3B cells were cultured, treated and assayed as in (A), except that 30 μM z-VAD-fmk or DMSO was added at the time of IFNB1 addition. Data represent mean ± SEM from four replicates and is representative of two independent experiments. Statistical analysis was performed using one-way ANOVA followed by Bonferroni’s multiple comparison test, *** = p < 0.001 compares DMSO-treated samples in the presence or absence of IFNB1 and ###p < 0.001 compares samples treated with IFNB1 in the presence of either DMSO or z-VAD-fmk. (E) Phase-contrast pictures of cells treated as in (D).
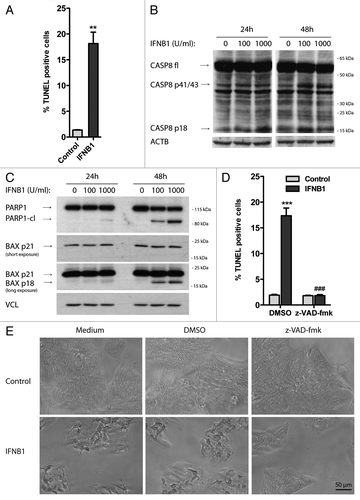
Figure 7. IFNB1 induced canonical autophagy in MCF-7 breast cancer cells and MEFs. (A) Knockdown efficiency of ATG7. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and transfected the next day with siRNA targeting ATG7 or the nonsilencing SCR control. Seventy-two hours post-transfection cells were lysed and western blot analysis performed with antibodies against ATG7 and VCL. (B) Silencing of ATG7 reduced basal, as well as IFNB1- and rapamycin-induced MAP1LC3B conversion. Cells were cultured and transfected as in (A). Forty-eight hours post-transfection, control medium, 1000 U/ml IFNB1 or 100 nM rapamycin was added for 24 h, before western blot analysis was performed for MAP1LC3B and ACTB protein levels. Relative expression levels are shown below the blot, which are representative of two independent experiments. (C) Atg7+/+ and Atg7−/− MEFs were IFNB1-responsive. Atg7+/+ and Atg7−/− MEFs were cultured for 24 h, serum starved for 3 h and treated with control medium or 100 or 1000 U/ml of IFNB1 for 30 min. Western blot analysis was performed for p-STAT, STAT, ATG7, ACTB and VCL protein levels. (D) IFNB1 induced MAP1LC3B conversion in MEFs. Atg7+/+ and Atg7−/− MEFs were cultured for 24 h and treated with control medium or 1000 U/ml IFNB1 for further 12 or 48 h. Western blots were performed for MAP1LC3B and VCL.
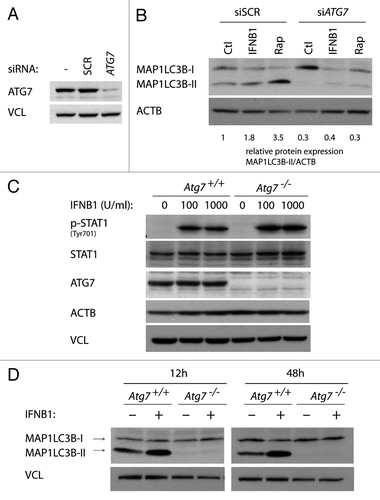
Figure 8. Blocking autophagy increased the proapoptotic activity of IFNB1. (A) Knockdown efficiency of core autophagy proteins. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and transfected the next day with siRNAs targeting ATG5, ULK1/2 or ATG7 as indicated. SCR was used as a nonsilencing control. Seventy-two hours post-transfection cells were lysed and western blot analysis performed with antibodies against ULK1, ATG5 and VCL. (B) Knockdown of core autophagy proteins reduced MAP1LC3B-II levels. MCF-7 eGFP-MAP1LC3B cells were cultured and transfected as in (A), but western blot analysis was performed for MAP1LC3B and ACTB levels. Relative expression levels are shown below the blot. (C) Knockdown of core autophagy proteins increased SQSTM1 levels. Cells were cultured and transfected as in (A), but western blot was performed for SQSTM1 and ACTB levels. Relative expression levels are shown below the blot. (D) IFNB1-induced autophagy did not modulate its antiproliferative response. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and transfected the next day with siRNAs targeting different core-autophagy proteins as indicated. Forty-eight hours post-transfection, control medium or 1000 U/ml IFNB1 was added for another 48 h before cell proliferation was measured by BrdU incorporation. Data represent mean and SEM of three independent experiments, each obtained from an average of four replicates. Statistical analysis was performed using one-way repeated measures ANOVA followed by Dunett’s post test against the SCR transfected sample, #p < 0.05 and ##p < 0.01 in the absence of IFNB1, while **p < 0.01 in the presence of IFNB1. Note the lack of significant interaction between IFNB1 treatment and any of the siRNA transfections using two-way repeated measures ANOVA. (E) IFNB1-induced autophagy counteracted its proapoptotic function. Cells were cultured, transfected and treated as in (A) and stained for DNA fragmentation with a TUNEL assay. Data represent mean and SEM of three independent experiments, where each value is an average of three replicates. The results were analyzed as in (A). #p < 0.05 and ***p < 0.001 using Dunett’s post test against the SCR transfected sample in the absence or presence of IFNB1 respectively. ¤p < 0.05, ¤¤p < 0.01 and ¤¤¤p < 0.001 indicate significant interaction between IFNB1 treatment and the indicated siRNA transfection using two-way repeated measures ANOVA. Significant interaction suggests a relatively larger effect of autophagy on the amount of TUNEL-positive cells in IFNB1-treated samples compared with control-treated samples.
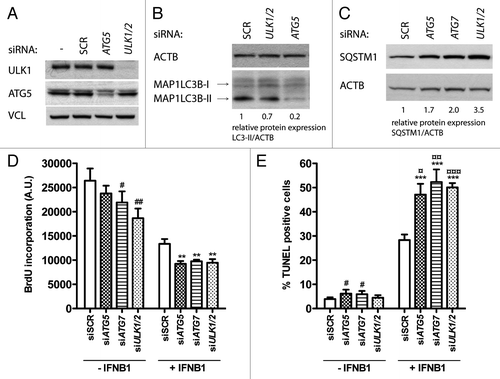
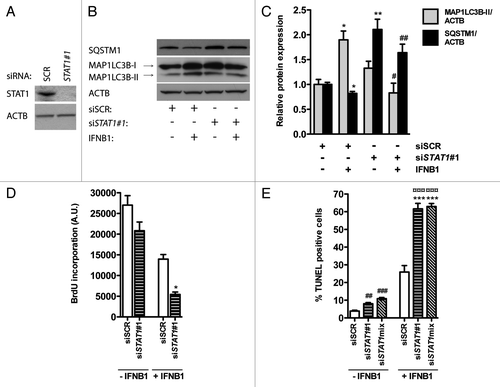
Figure 9. STAT1 was a positive regulator of IFNB1-induced autophagy. (A) Knockdown efficiency of STAT1. MCF-7 eGFP-MAP1LC3B cells were cultured for 24 h and transfected the next day with siRNAs targeting STAT1. SCR siRNA was used as a nonsilencing control. Seventy-two hours post-transfection cells were lysed and western blot analysis performed with antibodies against STAT1 and ACTB. (B) Silencing of STAT1 reduces IFNB1-induced autophagy. MCF-7 eGFP-MAP1LC3B cells were cultured and transfected as in (A). Forty-eight hours post-transfection control medium or 1000 U/ml IFNB1 was added for additional 24 h before cell lysis. Western blot analysis was performed with antibodies against SQSTM1, MAP1LC3B and ACTB. (C) Quantification of band intensities in (B). Data represent mean and SEM of three independent experiments. Statistical analysis was performed using Student’s t-test, *p < 0.05 or **p < 0.01 relative to SCR transfected samples in the absence of IFNB1; #p < 0.05 or ##p < 0.01 relative to SCR transfected samples in the presence of IFNB1. (D) STAT1-dependent autophagy did not affect IFNB1’s antiproliferative capacity. MCF-7 eGFP-MAP1LC3B cells were cultured and transfected as in (B), and 48 h post-transfection control medium or 1000 U/ml IFNB1 was added for additional 48 h before cell proliferation was measured by BrdU incorporation. Data represent mean and SEM of three independent experiments, each obtained from an average of three replicates. Statistical analysis was performed using Student’s paired t-test comparing SCR and STAT1 siRNA transfected samples, *p < 0.05 in the presence of IFNB1. Note the lack of a significant interaction between IFNB1 treatment and STAT1 siRNA silencing using two-way repeated measures ANOVA. (E) STAT1-dependent autophagy counteracted IFNB1’s proapoptotic function. MCF-7 eGFP-MAP1LC3B cells were cultured, transfected and treated as in (D) but stained for DNA fragmentation with a TUNEL assay. Data represent mean and SEM of four replicates and are representative of two independent experiments. Statistical analysis was performed using one-way ANOVA followed by Dunett’s post test against the SCR transfected sample, ##p < 0.01 and ###p < 0.001 in the absence of IFNB1, while ***p < 0.01 in the presence of IFNB1. ¤¤¤p < 0.001 indicates significant interaction between IFNB1 treatment and STAT1 silencing using two-way ANOVA. Significant interaction suggests a relatively larger effect of autophagy on the amount of TUNEL-positive cells in IFNB1-treated samples when compared with control-treated samples.