Figures & data
Figure 1. Overexpression of MIR181A resulted in decreased autophagic activity in MCF-7 cells. (A) MIR181A blocked starvation-induced GFP-LC3 dot formation in MCF-7 cells. Cells were cotransfected with MIR181A or control construct (MIR-CNT) together with GFP-LC3 plasmid and autophagy was assessed under no starvation or starvation (2 h) conditions. White arrows indicate clusters of the GFP-LC3 dots in cells. (B) Quantitative analysis of the experiments in (A). MIR181A overexpression, but not control (MIR-CNT) overexpression, blocked starvation-induced autophagy (mean ± SD of independent experiments, n = 3, **p < 0.01. N.S., not significant). (C) MIR181A decreased starvation-induced conversion of LC3-I to LC3-II in MCF-7 cells. Immunoblot results of extracts from nonstarved (STV-) or starved (STV+) cells (n = 3). LC3-II/LC3-I densitometric ratios are marked. ACTB was used as a loading control. (D) MIR181A blocked starvation induced SQSTM1 degradation in MCF-7 cells (n = 3). ACTB was used as a loading control. SQSTM1/ACTB densitometric ratios were marked. (E) MIR181A blocked rapamycin-induced GFP-LC3 dot formation. Cells were cotransfected with GFP-LC3 plasmid and MIR181A or MIR-CNT, treated with DMSO (carrier) or rapamycin (2.5 µM, 24 h). (F) Quantitative analysis of the experiments in (E) (mean ± SD of independent experiments, n = 4, *p < 0.05. N.S., not significant). (G) MIR181A decreased LC3-I to LC3-II conversion stimulated by rapamycin (RAP) in MCF-7 cells (n = 2). LC3-II/LC3-I ratios are marked. (H) MIR181A blocked rapamycin-induced SQSTM1 degradation in MCF-7 cells (n = 2).
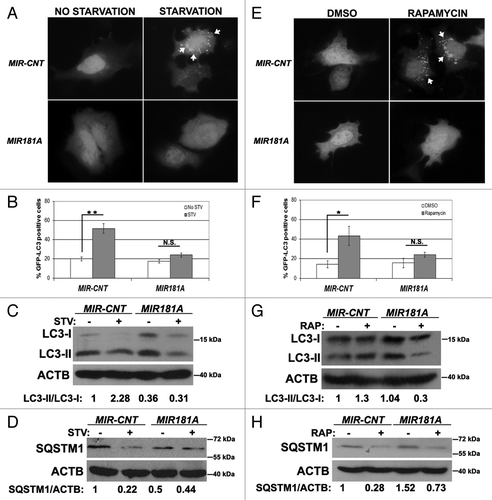
Figure 2. Effect of MI181A on autophagy is not cell-type dependent. (A) MIR181A blocked starvation-induced GFP-LC3 dot formation in Huh-7 cells. Cells were cotransfected with GFP-LC3 plasmid and MIR181A or MIR-CNT and autophagy was assessed under no starvation or starvation (4 h) conditions. (B) Quantitative analysis of the experiments in (A) (mean ± SD of independent experiments, n = 3, *p < 0.05. N.S., not significant). (C) MIR181A expression decreased starvation-induced LC3-I to LC3-II conversion in Huh-7 cells (n = 3). LC3-II/LC3-I ratios are marked. (D) MIR181A blocked starvation-induced SQSTM1 degradation in Huh-7 cells (n = 2). SQSTM1/ACTB ratios are marked. (E) MIR181A blocked rapamycin-induced GFP-LC3 dot formation. Autophagy was assessed following DMSO or rapamycin treatment (2.5 µM, 24 h). (F) Quantitative analysis of the experiments in (E) (mean ± SD of independent experiments, n = 3, **p < 0.01. N.S., not significant). (G) MIR181A resulted in decreased rapamycin-induced conversion of LC3-I to LC3-II in Huh-7 cells (n = 2). RAP, rapamycin. LC3-II/LC3-I ratios are marked. (H) MIR181A blocked rapamycin-induced SQSTM1 degradation in Huh-7 cells (n = 2). SQSTM1/ACTB ratios are marked.
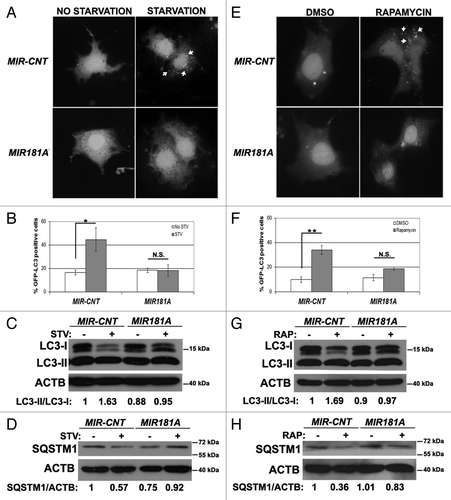
Figure 3. Inhibition of endogenous MIR181A using antagomirs (Ant-181a) stimulated autophagic activity. (A) Ant-181a enhanced starvation-induced GFP-LC3 dot formation. CNT-Ant, control antagomirs. (B) Quantitative analysis of the experiments in (A). NO STV, No starvation. STV, starvation for 20 min. (mean ± SD of independent experiments, n = 3, *p < 0.05, **p < 0.01). (C) Ant-181a, but not CNT-Ant stimulated starvation (STV, 4 h)-stimulated LC3-I to LC3-II conversion in MCF-7 cells (n = 4). LC3-II/LC3-I densitometric ratios are marked. (D) Ant-181a, but not CNT-Ant resulted in the stimulation of SQSTM1 protein degradation following starvation (4 h) in MCF-7 cells (n = 3). SQSTM1/ACTB ratios are marked.
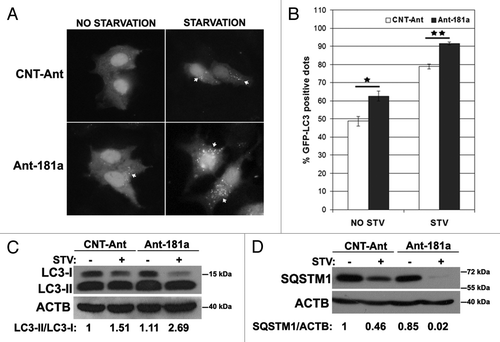
Figure 4.MIR181A affected ATG5 levels in MCF-7 cells. (A) MIR181A target sequence in the 3′ UTR of the ATG5 mRNA. The MIR181A seed sequence is marked in italics. (B) ATG5 protein levels were decreased following MIR181A overexpression in MCF-7 cells. Immunoblots of MIR-CNT or MIR181A transfected cells that were nonstarved (STV-) or starved (STV+) (n = 3). ACTB was used as a loading control. ATG5/ACTB band densitometric ratios are shown. (C) Quantitative PCR (qPCR) analysis of ATG5 mRNA levels in control (MIR-CNT) or MIR181A transfected MCF-7 cells (mean ± SD of independent experiments, n = 3, *p < 0.05). Data were normalized using GAPDH mRNA. (D) ATG5 protein levels were increased following antagomir-181a (Ant-181a) transfection. CNT-Ant, control antagomirs (n = 2). ATG5/ACTB ratios are shown.
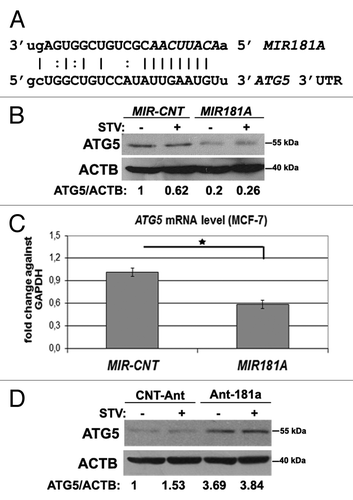
Figure 5. ATG5 overexpression rescued cells from MIR181A-mediated autophagy inhibition. MCF-7 cells were cotransfected with MIR181A or MIR-CNT and an ATG5 expression plasmid lacking the MIR181A target region. Autophagy was evaluated. (A) Immunoblot analysis of showing ATG5 and ACTB protein levels following indicated transfections (n = 2). ATG5/ACTB ratios are shown. (B) GFP-LC3 dot formation before or after starvation (2 h). ATG5, MIR181A-insensitive ATG5 expression plasmid. (C) Quantitative analysis of GFP-LC3 dot formation (mean ± SD of independent experiments, n = 3, **p < 0.01 and ***p < 0.001).
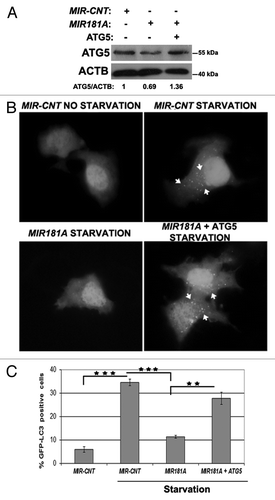
Figure 6. ATG5 as a direct target of MIR181A and changes in endogenous MIR181A levels during cellular stress. (A) A scheme representing luciferase constructs with wild-type (wt) or mutant 3′ UTR MIR181A MRE sequences of ATG5. Mutations are marked in lower case letters and underlined. (B) Normalized luciferase activity in lysates from 293T cells co-transfected with wild-type or mutant ATG5-luciferase constructs and MIR181A or MIR-CNT (mean ± SD of independent experiments, n = 3, **p < 0.01. N.S., not significant). (C) TaqMan quantitative PCR (qPCR) analysis of endogenous MIR181A levels under control (CNT, no starvation) or starvation (STV, 2 h and 4 h) conditions. Endogenous MIR181A levels were not responsive to starvation treatment (mean ± SD of independent experiments, n = 3, N.S., not significant). TaqMan qPCR data were normalized using U6 small nuclear 1 (RNU6-1) (U6) mRNA levels. (D) TaqMan qPCR analysis of endogenous MIR181A levels in cells treated with DMSO (carrier) or rapamycin. Endogenous MIR181A levels were increased following rapamycin treatment (mean ± SD of independent experiments, n = 4, *p < 0.05).
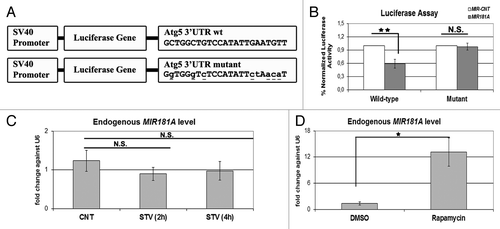
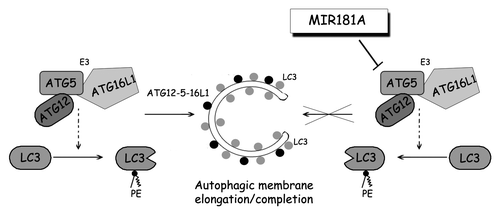
Figure 7. A model depicting the effect of MIR181A on autophagy pathways. MIR181A regulates stress-induced autophagy by targeting the key autophagy protein ATG5. The resulting decrease in ATG12–ATG5-ATG16L1 activity leads to attenuation of LC3 lipidation and inhibits the membrane elongation and completion stage of autophagy.