Figures & data
Figure 1. The process and regulation of autophagosome formation in mammalian cells. Mammalian autophagosome formation is induced by the activation of the ULK1 complex (comprising ULK1, ATG13, RB1CC1/FIP200 and C12orf44/ATG101), which is promoted by suppression of MTOR complex 1 (MTORC1) under starvation conditions or AMPK (both directly as well as by repressing MTORC1) in low-energy states. Once the activated ULK1 complex translocates to part of the endoplasmic reticulum (or possibly other intracellular membranes), it coordinates with the class III phosphatidylinositol-3-phosphate (PtdIns3P) kinase complex (including PIK3C3, BECN1, PIK3R4/p150, AMBRA1, UVRAG, ATG14 and SH3GLB1) for the initiation of autophagosomes upon formation of PtdIns3P and recruitment of double ZFYVE 1. Two ubiquitin-like systems including the ATG12–ATG5-ATG16L1 complex (containing ATG5, ATG7, ATG10, ATG12 and ATG16L1) and the LC3–phosphatidylethanolamine (PE) conjugation system (including ATG3, ATG4, ATG7 and MAP1LC3) will then promote the elongation of phagophore membranes for the formation of autophagosomes.
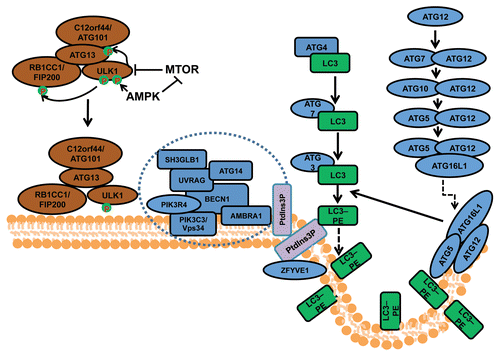
Figure 2. Autophagy in stemness of hematopoietic stem cells. During hematopoiesis, differentiated blood cells are generated from hematopoietic stem cells (HSCs). Long-term HSCs develop into short-term HSCs, multi- and then oligopotent progenitors and lineage-restricted progenitors, which give rise to the differentiated blood cells. These include erythrocytes, platelets, granulocytes, macrophages, dendritic cells and the lymphocytes T, B and NK cells. Autophagy is thought to play a role in self-renewal and quiescence in HSCs, the two hallmarks of stemness. Moreover, during differentiation autophagy is hypothesized to help multipotency and remodeling. Last, evidence is increasing that autophagy is required to maintain the healthy differentiated lineages.
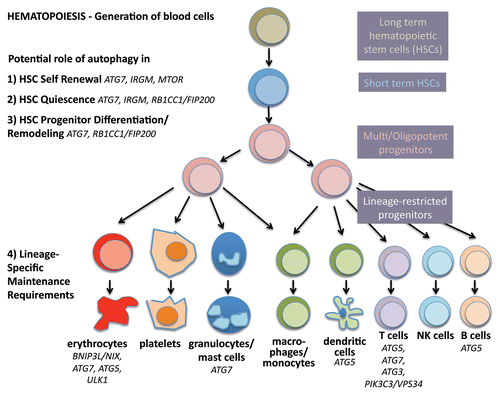
Figure 3. Autophagy/mitophagy and the CSC cellular state: Two models. Mitochondria can be eliminated through nonspecific autophagic processes or selectively removed by mitophagy. The former is generally related to metabolic responses that are imposed by a lack of nutrients. Mitophagy, however, permits a tight adjustment in the number of cellular mitochondria by regulating the selective degradation of damaged, dysfunctional and superfluous mitochondria and adjust to changing physiological demands. Mitophagy, therefore, can play a crucial role in adapting the number and quality of mitochondria to new microenvironmental conditions. (A) Cell-autonomous model. Upregulation of mitophagy leading to significant reductions in both the number and the size of mitochondria (i.e., the “mitochondrial phenotype” that is commonly associated with stem cells) concurrently enhances a bioenergetic shift from somatic oxidative mitochondria toward an alternative glycolytic phenotype (“direct Warburg effect”), an energetic infrastructure associated with the transcriptional networks responsible for stemness and pluripotency. (B) Non-cell-autonomous model (“autophagic tumor stroma model of cancer metabolism”). Cancer cells’ mitochondrial oxidative phosphorylation (OXPHOS) can induce oxidative stress in adjacent fibroblasts (cancer-associated fibroblasts [CAFs] via H2O2 and reactive oxygen species (ROS), resulting in the onset of a myofibroblastic autophagic phenotype in CAFs. The CAFs autophagic phenotype leads to a loss of mitochondria via mitophagy, forcing CAFs to undergo aerobic glycolysis (“reverse Warburg effect”). The products of aerobic glycolysis (such as L-lactate and ketones) are then reused by cancer cells for OXPHOS, resulting in increased mitochondrial mass in cancer cells. The utilization of the high-energy metabolites L-lactate and ketones in cancer cells can promote the transcriptional activation of CSC-like phenotypes.
![Figure 3. Autophagy/mitophagy and the CSC cellular state: Two models. Mitochondria can be eliminated through nonspecific autophagic processes or selectively removed by mitophagy. The former is generally related to metabolic responses that are imposed by a lack of nutrients. Mitophagy, however, permits a tight adjustment in the number of cellular mitochondria by regulating the selective degradation of damaged, dysfunctional and superfluous mitochondria and adjust to changing physiological demands. Mitophagy, therefore, can play a crucial role in adapting the number and quality of mitochondria to new microenvironmental conditions. (A) Cell-autonomous model. Upregulation of mitophagy leading to significant reductions in both the number and the size of mitochondria (i.e., the “mitochondrial phenotype” that is commonly associated with stem cells) concurrently enhances a bioenergetic shift from somatic oxidative mitochondria toward an alternative glycolytic phenotype (“direct Warburg effect”), an energetic infrastructure associated with the transcriptional networks responsible for stemness and pluripotency. (B) Non-cell-autonomous model (“autophagic tumor stroma model of cancer metabolism”). Cancer cells’ mitochondrial oxidative phosphorylation (OXPHOS) can induce oxidative stress in adjacent fibroblasts (cancer-associated fibroblasts [CAFs] via H2O2 and reactive oxygen species (ROS), resulting in the onset of a myofibroblastic autophagic phenotype in CAFs. The CAFs autophagic phenotype leads to a loss of mitochondria via mitophagy, forcing CAFs to undergo aerobic glycolysis (“reverse Warburg effect”). The products of aerobic glycolysis (such as L-lactate and ketones) are then reused by cancer cells for OXPHOS, resulting in increased mitochondrial mass in cancer cells. The utilization of the high-energy metabolites L-lactate and ketones in cancer cells can promote the transcriptional activation of CSC-like phenotypes.](/cms/asset/16f41840-49cf-489b-8c82-33b315c95b9d/kaup_a_10924132_f0003.gif)