Figures & data
Figure 1. A PGRMC1 ligand inhibits autophagic flux and increases MAP1LC3B-II. (A) In an autophagic flux assay, serum-fed (dashed lines) or serum-starved (solid lines) A549 cells were vehicle-treated (squares) or treated with 20 μM AG-205 (triangles) or 100 nM bafilomycin A1 (diamonds) for 16 h. Label was then “chased” for 4 h and samples were collected thereafter. Each point is derived from three independent samples, and the entire experiment is representative of four independent assays. The error bars indicate standard deviation. (B) LC3B localized by immunofluorescence to cytoplasmic puncta after AG-205 treatment for 24 h (lower panel). Scale bar: 20 μm. (C) Cells were treated with a vehicle control (lanes 1, 3, and 5) or with the PGRMC1 ligand AG-205 (lanes 2, 4, and 6, 20 μM). Cells were grown in 10% serum (lanes 1–2) or 0% serum without glutamine or glucose (lanes 3 to 6), and lysates were analyzed by western blot for LC3B (upper panels), SQSTM1 (middle panels), and GAPDH (lower panels). In lanes 5 and 6, cells were treated with 100 nM bafilomycin A1. With AG-205 treatment, LC3B-II increased, but there was no change in the levels of SQSTM1, a primary substrate of autophagy. The statistics and quantification for western is shown in Figure S1. (D) A549 cells were treated with 20 μM AG-205 (PGRMC1 ligand) for 0 to 72 h, resulting in increased LC3B-II. (E and F) NB-7 neuroblastoma cells were treated with 0, 50, 15, or 5 μM AG-205 (E) or haloperidol (F) and analyzed as for (C). (G) Electron microscopy showing A549 cells treated with vehicle control in 10% serum (top panel), vehicle control in 0% serum (middle panel), and AG-205 in 10% serum (lower panel). Scale bar: 10 μm. Overall, the results indicate that in multiple cell lines, PGRMC1 ligands initiate autophagy (indicated by LC3 cleavage) that is arrested (indicated by failure to degrade SQSTM1 and decrease starvation-induced increase amino acid release). Throughout, the results shown are representative of at least three independent experiments.
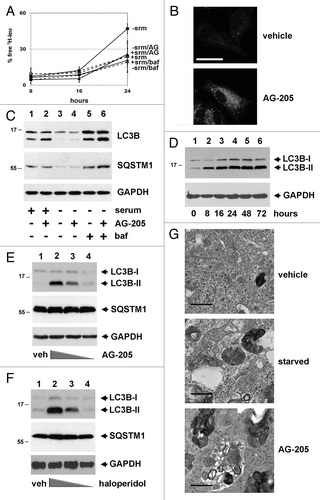
Figure 2.PGRMC1-knockdown disrupts autophagy. (A) Control (solid line, squares, “con”) and PGRMC1-knockdown (solid line, triangles, “shPGR”) A549 cells were analyzed for autophagic flux of 3H-leucine following 24 h of growth in media without serum and a 4 h “chase” period. PGRMC1-knockdown cells had a decrease in free 3H-leu release, which was not significantly different from control cells treated with 100 nM bafilomycin A1 (“baf”). Error bars represent standard deviation of triplicate measurements, and the data shown are representative of triplicate experiments. (B) Elevated MitoTracker Deep Red staining in PGRMC1-knockdown cells (upper right panel) compared with control cells (upper left panel). In the upper panel, the size bar represents 100 μm. Electron microscopy revealed an aberrant mitochondrial morphology in PGRMC1-knockdown cells (lower right panel), in which mitochondria have fewer christae compared with control cells (lower left panel). Scale bar: 1 μm. (C) PGRMC1-knockdown cells have elevated basal mitochondrial activity, (P = 0.006) measured as oxygen consumption rate (OCR, pmole/min/104 cells) but otherwise normal mitochondrial parameters. Error bars represent the standard deviation for five measurements, and the graph is representative of experiments performed in triplicate. Throughout the manuscript, *P ≤ 0.05; **P ≤ 0.01; and ***P ≤ 0.005. (D) LC3B, detected by immunofluorescence, was diffuse in control (top left) and punctate in PGRMC1-knockdown A549 cells (top right). Error bar: 20 μm. In the same cells, electron microscopy revealed increases in crescent-shaped autophagosomes (right, arrowheads) in PGRMC1-knockdown cells. Scale bars: 0.5 μm. (E) A549 control (lanes 1–3) or PGRMC1-knockdown (lanes 4 to 6) cells were maintained for 24 h in media with 10% serum (lanes 1 and 4) or DMEM without glutamine, glucose or serum (lanes 2 to 3 and 5 to 6). In lanes 3 and 6, 100 nM bafilomycin A1 was included. Lysates were analyzed by western blot for LC3B (top), SQSTM1 (middle), and GAPDH (bottom, loading control). LC3B-II increased in PGRMC1-knockdown cells grown in serum, while SQSTM1 levels were unaffected, and the difference in LC3B-II levels was suppressed by inhibiting autophagy. (F) Control (lanes 1–3) and PGRMC1-knockdown (lanes 4 to 6) A549 cells were transfected with a control plasmid (lanes 1 and 4), a plasmid encoding PGRMC1 (pRC40, lanes 2 and 5) or a plasmid encoding a heme-binding-deficient D120G mutant of PGRMC1 (pRC42, lanes 3 and 6). Lysates were analyzed for LC3B (top), SQSTM1 (middle), and GAPDH (bottom). LC3B-II was elevated in PGRMC1-knockdown cells and reversed by PGRMC1 (lane 5), but not the PGRMC1-hbd mutant.
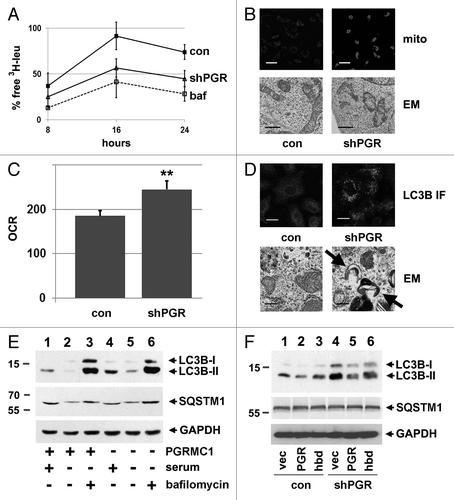
Figure 3. PGRMC1 associates with LC3B-II. (A) HEK293 human embryonic kidney cells were transfected with plasmid pRC40, encoding HA-tagged PGRMC1 (HA-PGR, lanes 1 and 2), or a control plasmid (lane 3), immunoprecipitated with an anti-HA epitope tag antibody and analyzed by western blot for HA-PGRMC1 (top panel) or LC3B (second panel). In lane 2, the cells were treated with 20 μM AG-205 for 24 h. The lower panels show PGRMC1 and LC3B levels in the lysates. (B) PGRMC1 (lane 1), the heme-binding deficient (hbd) D120G mutant of PGRMC1 (lane 2) or a control plasmid were expressed, precipitated and analyzed as in (A). (C) A549 non-small cell lung cancer cells were transfected with a control plasmid (lane 1) or the PGRMC1 expression plasmid pRC40 and treated with 20 μM AG-205 for 24 h. Lysates (lower panels) were immunoprecipitated with an anti-HA tag antibody and analyzed by western blot for LC3B (top) or HA-PGRMC1 (bottom). The lower band is due to the antibody light chain, which comigrates with PGRMC1. (D) LC3B was immunoprecipitated from A549 cells (lane 2) and probed for LC3B (top) or PGRMC1 (bottom). Lane 1 is an IgG control, and LC3B was weakly detectable. The input lysates are shown in the lower panels.
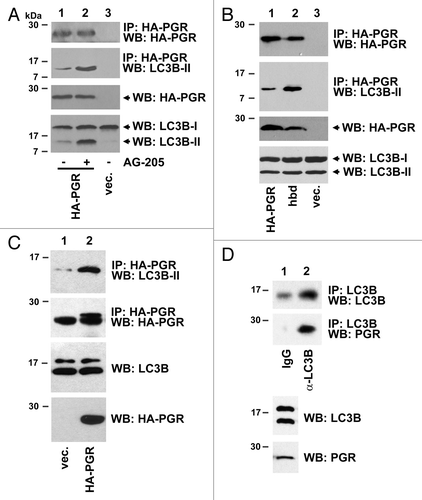
Figure 4. Endogenous PGRMC1 associates with LC3B. (A) A549 cells were immunoprecipitated with rabbit IgG or an equal amount of anti-PGRMC1 (α-PGR) antibody and analyzed for LC3B (top) or PGRMC1 (bottom). For (A and B), the lysates used in the precipitation were analyzed by western blot (lower panels) as indicated. (B) The same experiment as (A) was performed with lysates from HEK293 cells, where precipitates were analyzed for LC3B (top) or PGRMC1 (bottom), and the input lysates were measured in the two lower panels. (C) PGRMC1 (left) and LC3B (center) were analyzed by immunofluorescence in A549 cells starved for 24 h, and the proteins colocalized (right) in cytoplasmic vesicles. Scale bar: 20 μm. (D) PGRMC1-GFP (left) and LC3B-mCherry (right) were coexpressed in A549 cells and colocalized (right) in cytoplasmic puncta. Scale bar: 20 μm.
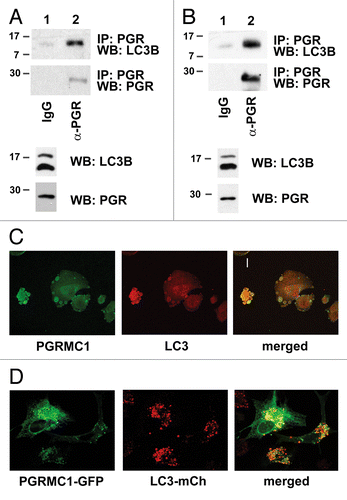
Figure 5. PGRMC1 inhibits AMPK signaling. (A) Kinase signaling intermediates in control (gray bars) and PGRMC1-knockdown A549 cells (shPGR, black bars) were screened using proteome profiler antibody array, and significant changes in the MTOR pathway are indicated. Error bars represent standard deviation of four measurements, and the array profiling was performed in duplicate. Cells were serum-starved for 24 h prior to this analysis and for the western blots in (B and C). (B) Decreases in MTOR signaling were verified by western blot analysis, showing decreased MTOR-Ser2448 and RPS6KB-Thr421 phosphorylation in PGRMC1-knockdown cells. (C) Increased AMPK-Thr174 phosphorylation in PGRMC1-knockdown cells was verified by western blot (second panel), which also revealed decreased AMPK levels (top) and increased TSC1 and TSC2 levels (lower panels), which are downstream of AMPK. The samples in (B and C) are identical, so the same loading control is shown for each. (D) Quantification and statistical analysis of the results from (C), measured in duplicate as band intensity relative to the loading control.
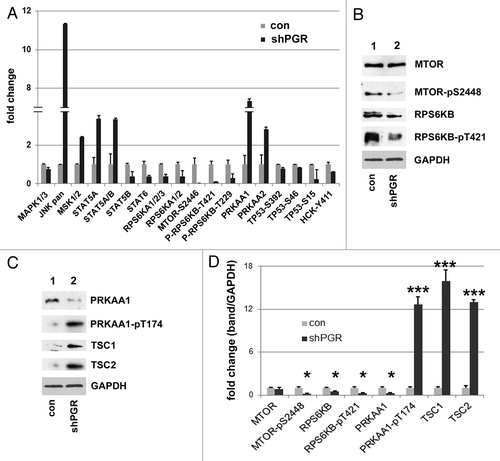
Figure 6. PGRMC1 promotes the degradation of ubiquitinated proteins. (A) LC3B was immunoprecipitated from control cells (lane 2) or PGRMC1-knockdown cells (shPGR, lane 3). Lane 1 is an identical precipitation reaction of control cell lysates (identical to lane 2) with a control antibody. Precipitates were analyzed for LC3B (top), SQSTM1 (second panel) or TUBA (third panel). (B) The loading controls for the source lysates for (A) revealed little change in SQSTM1 and TUBA levels. (C) Lysates from control (lane 1) or PGRMC1-knockdown cells (lane 2) were probed by western blot for ubiquitinated proteins. In lanes 3 and 4, A549 cells were treated with a vehicle (lane 3) or 20 μM AG-205 (lane 4) for 24 h. (D) Control cells (lanes 1 to 3) or PGRMC1-knockdown cells (lanes 4 to 6) were grown in serum-containing media, treated with vehicle (lanes 1 and 4), 30 μM chloroquine (lanes 2 and 5) or 100 nM bafilomycin A1 (lanes 3 and 6) for 24 h and analyzed for LC3B (top), ubiquitin (middle panel) or TUBA (lower panel). (E) A549 cells were treated with vehicle (lanes 1–3) or 20 μM AG-205 (lanes 4 to 6) and bafilomycin A1 or chloroquine, as described for (D) and analyzed by western blot as for (D).
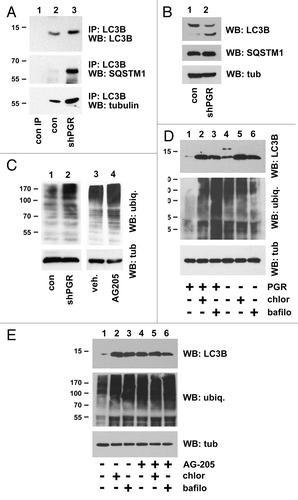
Figure 7. PGRMC1 associates with UVRAG. (A) HEK293 cells were transfected with a control plasmid (lane 1), pRC40, encoding PGRMC1 (PGR, lanes 2 and 3), or a plasmid encoding heme-binding deficient PGRMC1 (hbd, lane 4). In lane 3, cells were treated with 20 μM AG-205 for 24 h. PGRMC1 was immunoprecipitated and analyzed by western blot for PGRMC1 (top panel) or UVRAG (second panel). The lower panels show PGRMC1 and UVRAG levels, respectively, in the lysates. (B) PGRMC1 (lane 2) was immunoprecipitated from UVRAG-transfected A549 cells and probed for UVRAG (top panel) or PGRMC1 (lower panel). Lane 1 shows an identical precipitation reaction with preimmune serum from the same animal (PIS). The offset panel shows UVRAG expression in the lysate. (C) UVRAG fused to a flag epitope tag was expressed in A549 cells, immunoprecipitated and analyzed by western blot for UVRAG (top panel) or PGRMC1 (lower panel). (D) The input loading control is shown for (C).
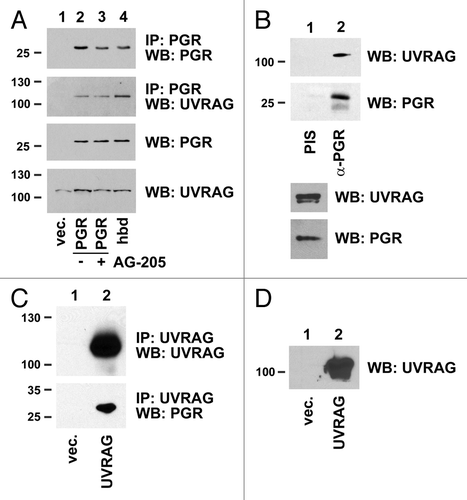
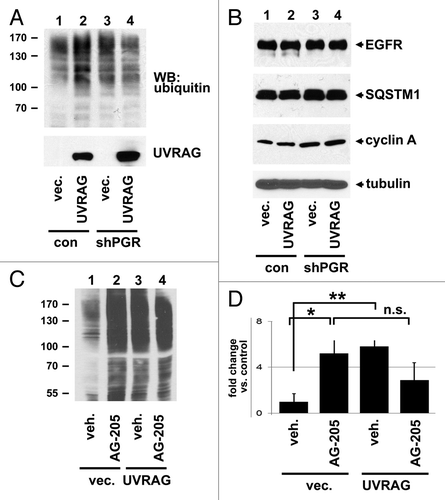
Figure 8. PGRMC1 counteracts UVRAG in regulating ubiquitinated protein levels. (A) Control (lanes 1 and 2) and PGRMC1-knockdown A549 cells (shPGR, lanes 3 and 4) were transfected with a control plasmid (lanes 1 and 3) or a UVRAG expression vector (lanes 2 and 4), maintained in serum, and analyzed by western blot for ubiquitin (top) or UVRAG (bottom). (B) The lysates from (A) were analyzed by western blot for EGFR, SQSTM1, CCNA1/cyclin A1, and TUBA. (C) A549 cells were transfected with a control plasmid (lanes 1 and 2) or a UVRAG expression vector (lanes 3 and 4) and treated with either vehicle (lanes 1 and 3) or the PGRMC1 ligand AG205 at 20 μM (lanes 2 and 4). Samples were then analyzed for ubiquitin. (D) Quantification of triplicate experiments described in (C), where the error bars represent standard deviation of triplicate measurements. The experiment was performed in quadruplicate. The results suggest that PGRMC1 associates with UVRAG and that UVRAG requires PGRMC1 to increase levels of ubiquitinated proteins.