Figures & data
Figure 1 PKM1 and PKM2 are highly expressed in the tumor stroma of human breast cancer samples lacking stromal Cav-1. Human breast cancer samples were immuno-stained with antibodies against Cav-1, PKM1, and PKM2. Antibodies directed against PKM1 and PKM2 are isoform-specific. Upper left panel: a representative example of a loss of Cav-1 staining in stromal cells is shown. Loss of Cav-1 expression occurs selectively in cancer-associated fibroblasts, but Cav-1 expression is retained in endothelial cells. Arrows indicate Cav-1 positive vessels. Right panels: PKM2 staining was performed using two different antibodies on human breast cancers lacking stromal Cav-1. Note that PKM2 expression is increased in the tumor stroma and displays two expression patterns. Upper right: PKM2 expression (detected with a Cell Signaling antibody) is largely confined within the stromal cells. Lower right: PKM2 (detected with a Proteintech antibody) can be also detected in the extracellular matrix of human breast cancers. Lower left panel: note that PKM1 is also upregulated in the stroma of human breast cancer samples lacking stromal Cav-1. Original magnification for all, 40×.
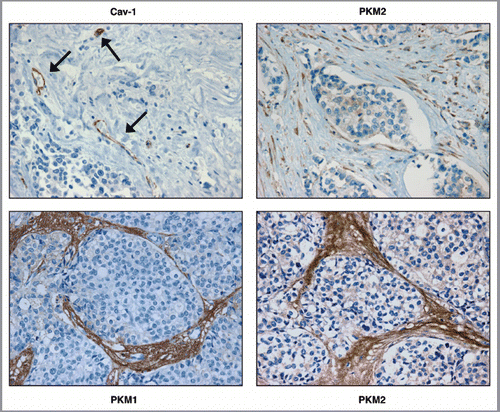
Figure 2 Overexpression of PKM1 and PKM2 in human fibroblasts. Human immortalized fibroblasts (hTERT-BJ1) were transduced with empty vector (EV), PKM1 or PKM2 lentiviral particles. After selection with puromycin, the cellular pools were subjected to immunoblot analysis to confirm PKM1 and PKM2 overexpression. Immunoblotting with β-actin is shown as an equal loading control.
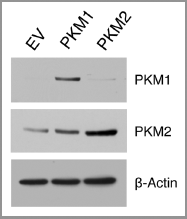
Figure 3 PKM1 induces increased lactate production in fibroblasts. EV, PKM1 or PKM2 fibroblasts were grown for 48 h in media supplemented with 2% FBS. Then, the media was collected and subjected to a biochemical analysis to determine lactate content. The graph shows that PKM1 overexpressing fibroblasts display a 1.5-fold increase in lactate secretion, as compared with control empty vector (EV) cells. In contrast, PKM2 overexpressing fibroblasts show similar lactate production as control cells. p = 0.04.
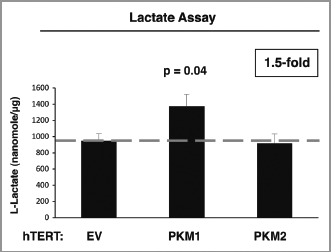
Figure 4 PKM2 overexpression induces NFκB-dependent autophagy in fibroblasts. (A) Western blot. Cells were analyzed by immuno-blot analysis using antibodies directed against a panel of autophagy markers. Note that fibroblasts harboring PKM2 show increased expression of the autophagy markers BNIP3, Beclin-1, and Cathepsin B (active form). Conversely, PKM1 did not induce autophagy. β-actin was used as equal loading control. (B) Immunofluorescence. After 24 h of serum starvation, cells were fixed and immuno-stained with antibodies against Beclin-1, BNIP3 and BNIP3L (all red). DAPI was used to visualize nuclei (blue). Note that PKM2 overexpression promotes an autophagic program in fibroblasts. No effects were observed in PKM1 overexpressing cells. (C) PKM2 induces NFκB activation. To gain insights into the molecular drivers of increased autophagy in PKM2 fibroblasts, cells were analyzed by immuno-blot analysis using antibodies directed against phospho-NFκB and HIF1α. Note that fibroblasts overexpressing PKM2 show increased activation and expression levels of NFκB. Conversely, NFκB is not activated in PKM1 cells. In addition, PKM1 and PKM2 do not stabilize HIF1α expression in fibroblasts. β-actin was used as equal loading control. (D) PKM2-induced autophagy is NFκB-dependent. PKM2 overexpressing fibroblasts were treated with increasing concentration of the NFκB inhibitor PS-1145 for 24 h. Then, cells were analyzed by immuno-blot analysis with antibodies against BNIP3. Note that NFκB inhibition reduces the expression levels of BNIP3 (an autophagy marker) in a dose-dependent manner. β-actin was used as equal loading control.
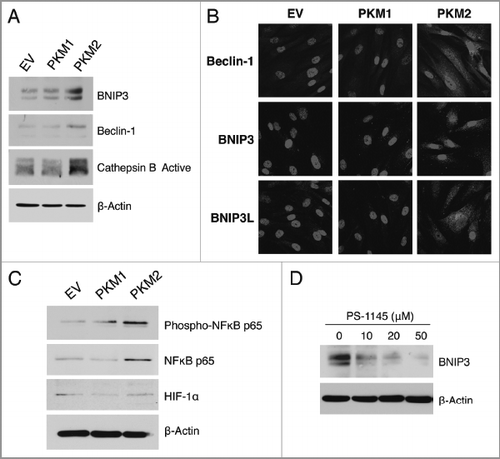
Figure 5 Stromal expression of PKM2 promotes ketone body metabolism. (A) β-hydroxybutyrate assay. The β-Hydroxybutyrate (β-OH-butyrate) concentration was measured in the cell culture media of EV, PKM1 and PKM2 fibroblasts, and was normalized for protein concentration. Note that fibroblasts overexpressing PKM2 display a 1.4-fold increase in β-Hydroxybutyrate accumulation relative to control empty vector (EV) fibroblasts. Fibroblasts overexpressing PKM1 show β-Hydroxybutyrate production as control cells. (B) Western blot analysis. Fibroblasts expressing EV, PKM1 or PKM2 were analyzed by immuno-blot with a panel of antibodies against key enzymes of ketone body metabolism (both production and utilization). Note that PKM2-fibroblasts show elevated expression of the enzymes involved in ketone production (HMGCS1, HMGCS2 HMGCL, and BDH1), and ketone utilization (OXCT1 and OXCT2). Conversely, expression of PKM1 in fibroblasts abrogates the expression of enzymes involved in ketone production (HMGCS1, HMGCS2), and ketone utilization (OXCT1 and OXCT2) relative to EV-fibroblasts. These data indicate that ketone body metabolism is activated by PKM2, but abolished by PKM1. β-actin was used as equal loading control.
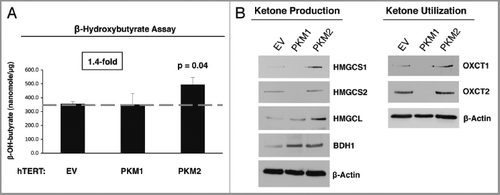
Figure 6 Fibroblasts overexpressing PKM1 and PKM2 promote mitochondrial activity in adjacent cancer cells. MitoTracker staining was used to visualize mitochondrial activity in homotypic cultures and in co-cultures of fibroblasts and GFP-positive MDA-MB-231 cells. DAPI was used to visualize nuclei (blue). (A) Fibroblast homotypic cultures. Note that fibroblasts expressing PKM1 or PKM2 exhibit high levels of MitoTracker staining (red) relative to empty vector (EV) control cells, consistent with increased mitochondrial activity. (B) Co-cultures. Note that fibroblasts expressing PKM1 or PKM2 increase the MitoTracker staining (red) of adjacent GFP-positive MDA-MB-231 cells (green), relative to empty vector (EV) control cells. In the top panels, only the red channel is shown, to appreciate the increased MitoTracker staining of cancer cells. The white stars identify the nuclei of GFP positive MDA-MB-231 cells. In addition, PKM1 and PKM2 fibroblasts co-cultured with cancer cells show reduced MitoTracker staining compared with respective homotypic cultures (compare panel A and B).
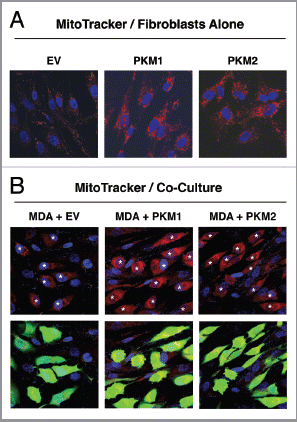
Figure 7 PKM1 and PKM2 overexpressing fibroblasts do not induce apoptosis in co-cultured breast cancer cells. GFP-positive MDA-MB-231 cells were co-cultured either with EV, PKM1 or PKM2 fibroblasts. Corresponding homotypic cultures were established in parallel. To measure apoptosis, cells were subjected to Annexin-V staining and analyzed by FACS. Thus, the GFP (+) and GFP (−) cells represent breast cancer cells and fibroblasts, respectively. (A) Apoptotic rates of co-cultured MDA-MB-231 cells. MDA-MB-231 cells maintained in co-culture with EV control fibroblasts show a 7-fold reduction in apoptosis, as compared with MDA-MB-231 cells cultured alone. Interestingly, MDA-MB-231 cells co-cultured either with PKM1 or PKM2 fibroblasts display a similar protection against apoptosis, indicating that PKM1 and PKM2 fibroblasts do not impart any toxicity effects. (B) Apoptotic rates of co-cultured fibroblasts. When maintained in co-culture with MDA-MB-231 cells, EV, PKM1 or PKM2 fibroblasts display an up to 2-fold reduction in apoptosis.
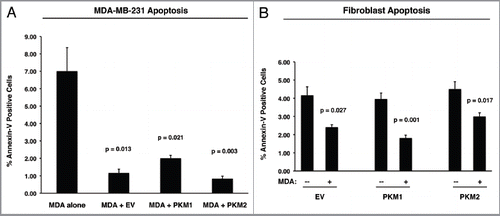
Figure 8 Fibroblasts overexpressing PKM1 and PKM2 promote breast cancer tumor growth, with decreased angiogenesis. (A) Tumor growth. We used a xenograft model employing MDA-MB-231 breast cancer cells co-injected with either EV, PKM1 or PKM2 fibroblasts into the flanks of athymic nude mice. Tumor weights and volumes were measured at 4 weeks post-injection. Note that fibroblasts overexpressing PKM2 greatly promote tumor growth, resulting in a 2-fold increase in tumor weight, and a 2.4-fold increase in tumor volume. Similarly, fibroblasts overexpressing PKM1 augment tumor growth, inducing a 1.5-fold increase in tumor weight, and a 2-fold increase in tumor volume. P values are as shown. n = 10 tumors per experimental group. (B and C) Tumor Angiogenesis. Tumor frozen sections were cut and immuno-stained with anti-CD31 antibodies. (B) Vascular density (number of vessels per field) quantification. Note that a significant decrease (1.3- to 1.5-fold) in vessel density was observed, suggesting that the tumor promoting effects of fibroblasts harboring PKM1 or PKM2 are independent of angiogenesis. P values are as shown. (C) Representative images of CD31 immuno-staining. Original magnification, 40×.
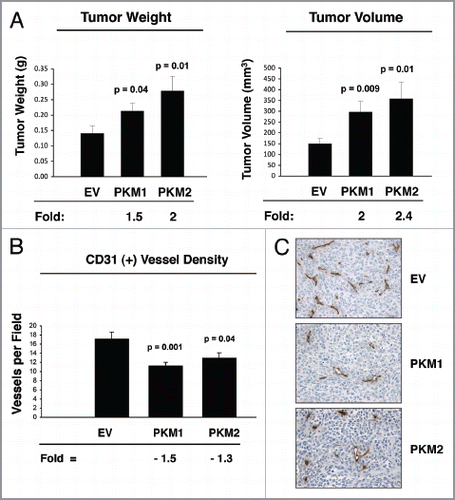
Figure 9 Fibroblasts overexpressing PKM1 promote an inflammatory response in tumor tissue. To evaluate inflammation, frozen sections of the tumor xenograft were analyzed using CD45 antibodies, detecting nucleated hematopoietic cells, including neutrophils and macrophages. Note that CD45 is highly expressed in PKM1-tumors, indicative of a significant increase in the inflammatory response. Original magnification, 40×.
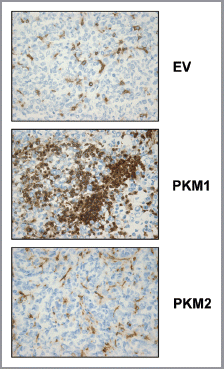
Figure 10 Fibroblasts overexpressing PKM2 induce oxidative mitochondrial activity in breast cancer cells in vivo. To evaluate mitochondrial activity in situ, the activity of Complex IV was detected via COX (Cytochrome C Oxidase) activity staining of frozen sections derived from tumor xenografts. Note that COX activity is increased specifically in the cancer cells of PKM2-tumors. Conversely, PKM1-tumors do not display increased mitochondrial activity. Original magnification, 60×.
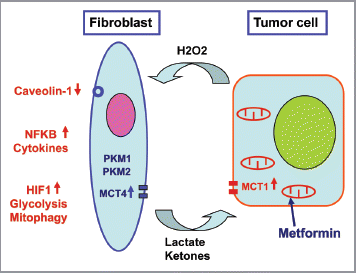
Figure 11 Pyruvate kinase expression (PKM1 and PKM2) in cancer associated fibroblasts drives tumor growth. Cancer cells secrete hydrogen peroxide (H2O2) which initiates oxidative stress in adjacent stromal fibroblasts. This, in turn, drives the activation of two main transcription factors, namely HIF1 and NFκB. NFκB is the master regulator of the innate immune response, resulting in the secretion of inflammatory cytokines. HIF1-α stabilization induces autophagy, mitophagy, and aerobic glycolysis. As a consequence, PKM1 and PKM2 expression results in the production of excess L-lactate and ketone bodies (3-hydroxy-butyrate) that are extruded via a mono-carboxylate transporter, known as MCT4. These high-energy nutrients are then taken up and recycled by cancer cells using another mono-carboxylate transporter, namely MCT1. Thus, L-lactate and ketones produced by stromal fibroblasts are transferred to cancer cells and used as fuel for mitochondrial oxidative metabolism (OXPHOS), producing large amounts of ATP. This model explains the anti-cancer activity of Metformin, a diabetic drug, that mechanistically functions as a mitochondrial “poison,” as it is a specific inhibitor of mitochondrial complex I. Complex I is a key part of the electron transport system for generating ATP. Note also that a loss of stromal Caveolin-1 (Cav-1) occurs, as it is degraded by stromal autophagy. Loss of stromal Cav-1 further perpetuates oxidative stress, and is a powerful biomarker for predicting early tumor recurrence, metastasis, and drug-resistance, as well as poor clinical outcome in breast cancer patients.