Figures & data
Figure 1. MEK inhibitors interact with CHK1 inhibitors to kill multiple primary human glioma cell isolates. (A) GBM5 and GBM6 cells were treated with AZD7762 (100 or 300 nM), CI-1040 (0.5 or 1.0μM), or AZD7762+CI-1040 for 24 h. Floating and attached cells were isolated after drug exposure and cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value. (B) GBM5 cells were treated with UCN-01 (10 or 50nM), AZD7762 (100 or 300 nM), CI-1040 (0.5 or 1.0μM), AZD6244 (0.5 or 1.0μM) or UCN-01+CI-1040 or AZD7762+AZD6244 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value; Upper blot: portions of the cell lysates were immunoblotted against cleaved caspases-3, and cleaved caspases-7. (C) GBM6 cells were treated with UCN-01 (10, 25, or 50 nM), CI-1040 (0.5 or 1.0μM), AZD6244 (0.5, 1.0 μM) or UCN-01+CI-1040 or AZD6244+UCN-01 for 24 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value. (D) GBM6 cells were treated with UCN-01 (10–50nM), AZD7762 (100 or 300 nM), CI-1040 (0.5 or 1.0 μM), UCN-01+CI-1040 or AZD7762+CI-1040 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value. (E) GBM12 and GBM14 cells were treated with UCN-01 (10 or 50 nM), CI-1040 (0.5 or 1.0μM) or UCN-01+CI-1040 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value. (F) GBM5, 6, 12, and 14 cells were treated with AZD7762 (300 nM), AZD6244 (500 nM) or AZD7762+AZD6244 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value.
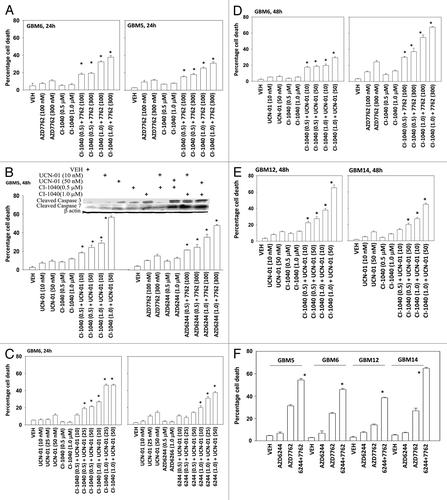
Table 1. Pre-treatment of GBM6 cells with inhibitors of MEK1/2 and CHK1 causes radiosensitization. GBM6 cells, plated as single individual cells in sextuplicate, were treated with AZD6244 (500 nM); AZD7762 (100 nM) or the drug combination for 48h. The drug containing media was removed and fresh media lacking drugs added to the cultures. Cells were then irradiated (1 Gy, 2 Gy) and colonies permitted to form over the following 14 d. Data are the means of all conditions from multiple experiments (n = 3, ± SEM). #p < 0.05 less than CHK1 inhibitor value
Table 2. Concomitant drug treatment and radiation exposure radiosensitizes GBM6 cells. GBM6 cells, plated as single individual cells in sextuplicate, were treated with AZD6244 (500 nM); AZD7762 (100 nM) or the drug combination. Fifteen minutes after drug treatment cells were irradiated (1 Gy, 2 Gy). Forty eight h after drug treatmentthe drug containing media was removed and fresh media lacking drugs added to the cultures. Colonies permitted to form over the following 14 d. Data are the means of all conditions from multiple experiments (n = 3, ± SEM). #p < 0.05 less than CHK1 inhibitor value
Figure 2. MEK inhibitors interact with CHK1 inhibitors to kill human kill pediatric medulloblastoma cell lines. Medulloblastoma cell lines (A) DAOY (B) VC312; and (C) D283 cells were treated with UCN-01 (50nM), AZD7762 (300 nM), CI-1040 (1.0 μM), AZD6244 (500 nM), UCN-01+CI-1040, AZD7762+CI-1040 or AZD7762+AZD6244 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value.
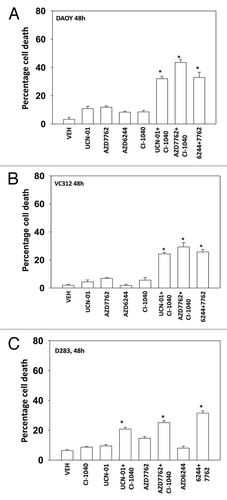
Figure 3. MEK + CHK1 inhibitors cause glioma and medulloblastoma cell death that is associated with dephosphorylation of ERK1/2 and phosphorylation of JNK and p38. (A) and (B) GBM5, 6 and 14 cells were treated with AZD7762 (25 or 50 nM), AZD6244 (500 nM) or AZD7762+AZD6244 for 24 h. Cell lysates were immunoblotted against cleaved caspase-3, PARP1, P-ERK1/2, P-JNK1–3, P-p38, and P-S6. (C) GBM5 and 14 cells were treated with UCN-01 (10 or 50nM), CI-1040 (0.5 or 1.0 mM) or UCN-01+CI-1040 for 24 h. The cell lysates were immunoblotted against P-ERK1/2, P-p38 and P-JNK1–3. (D) DAOY cells were treated with UCN-01 (50 nM), AZD7762 (50 nM), AZD6244 (500 nM), CI-1040 (1.0 μM), UCN-01+CI-1040, AZD7762+CI-1040, or AZD7762+AZD6244 for 24 h. Cell lysates were immunoblotted against caspase-3, cleaved caspase-7, PARP1, P-JNK1–3, P-S6, P-ERK1/2 and P-AKT.
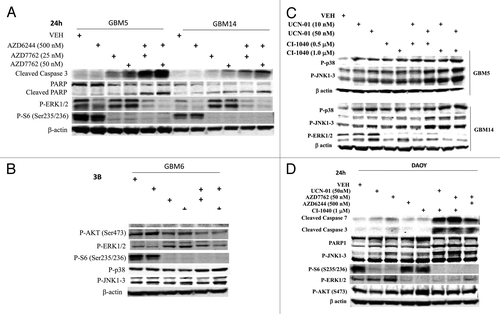
Figure 4. Inactivation of MEK/12 and p70 S6K and activation of JNK1–3/p38 and BAX/BAK are essential for MEK + CHK1 inhibitor killing in glioma and medulloblastoma cells. (A) GBM5, GBM12 and GBM14 cells were transfected with plasmids to express constitutively active p70 S6K, active MEK1 or empty vector CMV control. Twenty four hours after transfection, the cells were treated with PD98059 (25.0 μM) and AZD7762 (300 nM) for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) # p < 0.05 less than CHK1 inhibitor + MEK1/2 inhibitor value. (B) GBM5 and 14 cells were infected using a recombinant adenovirus to express dn-p38 in the presence or absence of JNK inhibitory peptide (JNK-IP, 10 μM). Thirty six h after transfection, the cells were treated with CI-1040 (1.0 μM), UCN-01 (50nM) or CI-1040+UCN-01 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) # p < 0.05 less than CHK1 inhibitor + MEK1/2 inhibitor value. (C) Top: GBM6 cells were treated with AZD7762 (300 nM) + AZD6244 (500 nM) for 24 or 48 h. Lysates were isolated for blotting of the indicated proteins and for immunoprecipitation of activated BAX and activated BAK. Bottom: GBM6 cells were infected with dn-p38 and 24 h after infection, the cells were treated with AZD7762 (300 nM) + PD98059 (25.0 μM) for 48 h in the presence or absence of JNK-IP (10 uM). The activity of BAK and BAX was determined after immunoprecipitation of the conformationally active BAX and BAK proteins. (D) GBM5 and GBM6 cells were infected with BCL-XL or empty vector control CMV. Twenty four h after transfection, the cells were treated with AZD7762 (300 nM) + AZD6244 (500 nM) in the presence or absence of HA14–1 (HA14–1, 500 nM) for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) # p < 0.05 less than CHK1 inhibitor + MEK1/2 inhibitor value.
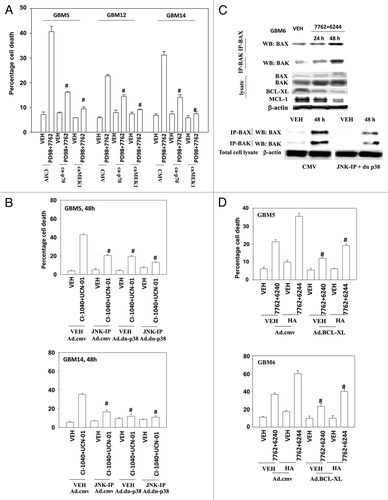
Figure 5. A SRC kinase inhibitor interacts with CHK1 inhibitors to kill primary human glioma cell isolates and pediatric medulloblastoma cell lines. (A) GBM6 and GBM12 cells and (B) GBM5 and GBM14 cells were treated with AZD7762 (300 nM), AZD0530 (100 nM), or AZD7762+AZD0530 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion based cell staining (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value. (C) GBM5 and GBM14 cells were treated with AZD7762 (300 nM), AZD0530 (100 nM), or AZD7762+AZD0530 for 24 h and cell lysates were immunoblotted against cleaved caspase 3, PARP1 and P-ERK1/2. (D) Medulloblastoma cell lines DAOY, VC312 and D283 were treated with AZD7762 (300 nM), AZD0530 (100 nM), or AZD7762+AZD0530 for 48 h. Floating and attached cells were isolated after drug exposure, cell viability was measured by trypan blue exclusion (± SEM, n = 3) * p < 0.05 greater than CHK1 inhibitor value. The cell lysates were immunoblotted against cleaved caspases-3, cleaved caspases-7 and P-ERK1/2.
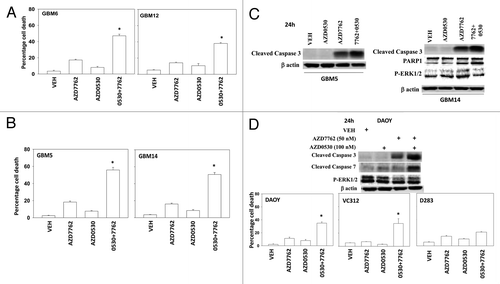
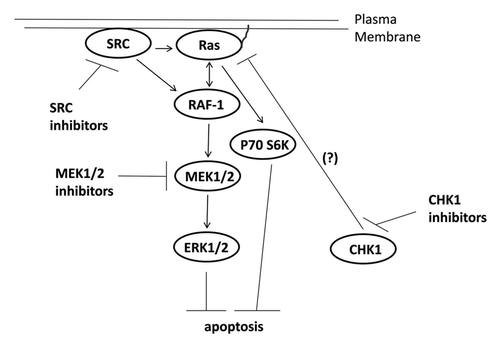
Figure 6. Possible signaling pathways by which CHK1 inhibitors activate ERK, and mechanisms by which MEK1/2 and SRC inhibitors potentiate CHK1 inhibitor lethality. CHK1 inhibitors through mechanisms not fully understood causes activation of the ERK1/2 pathway downstream of RAS. SRC kinases can increase ERK1/2 activity both by promoting RAS activation and also downstream of RAS at the level of RAF-1 tyrosine phosphorylation. Inhibition of either MEK1/2 disrupts this activation of ERK1/2 leading to tumor cell death.