Figures & data
Figure 1. The HaCaT cell system: Immortalized and transformed with oncogenes. HaCaT cells are immortalized epidermal keratinocytes, but they are not transformed, and they do not form tumors in immunodeficient mice. However, overexpression of activated H-Ras (G12V) or NFkB (p65 subunit) drives cell transformation, and confers the capacity for tumor cell engraftment, leading to tumor formation in nude mice. Thus, phenotypic comparisons of these 3 matched cell lines should allow one to better understand the metabolic requirements for successful tumor cell engraftment within the naïve host microenvironment.
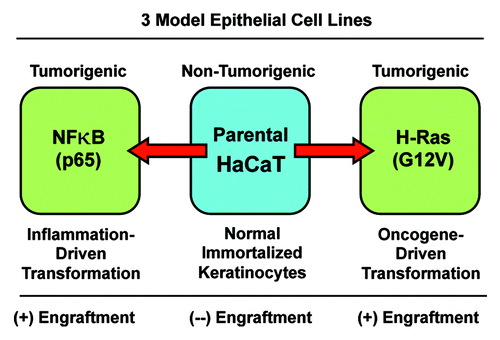
Figure 2. HaCaT-fibroblast co-cultures: Transformed HaCaT cells show a modest increase in ROS production, without any significant increases in glucose uptake. (A) ROS-production. (B) Glucose uptake. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 4 days with hTERT-immortalized fibroblasts (RFP[+]). Then, ROS production (a measure of oxidative stress) and glucose uptake (a measure of glycolytic activity) in HaCaT cells were quantitatively determined by FACS sorting. Note that only HaCaT-Ras cells show a significant increase in ROS production (1.5-fold; P = 0.03), without any detectable increases in glucose uptake.
![Figure 2. HaCaT-fibroblast co-cultures: Transformed HaCaT cells show a modest increase in ROS production, without any significant increases in glucose uptake. (A) ROS-production. (B) Glucose uptake. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 4 days with hTERT-immortalized fibroblasts (RFP[+]). Then, ROS production (a measure of oxidative stress) and glucose uptake (a measure of glycolytic activity) in HaCaT cells were quantitatively determined by FACS sorting. Note that only HaCaT-Ras cells show a significant increase in ROS production (1.5-fold; P = 0.03), without any detectable increases in glucose uptake.](/cms/asset/9fa14b29-2ea5-4502-b932-dd64b7a8fe03/kccy_a_10925510_f0002.gif)
Figure 3. HaCaT-fibroblast co-cultures: Cancer-associated fibroblasts show dramatic increases in both ROS production and glucose uptake. (A) ROS-production. (B) Glucose uptake. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 4 days with hTERT-immortalized fibroblasts (RFP[+]). Then, ROS production (a measure of oxidative stress) and glucose uptake (a measure of glycolytic activity) in hTERT-fibroblasts were quantitatively determined by FACS sorting. Note that hTERT-fibroblasts co-cultured with HaCaT-Ras cells show a significant increase in both ROS production (2.5-fold; P = 0.008) and glucose uptake (2.2-fold; P = 0.03). Similarly, hTERT-fibroblasts co-cultured with HaCaT-p65 cells show a significant increase in ROS production (1.9-fold; P = 0.01) and glucose uptake (1.7-fold; P = 0.02). Thus, oncogene-transformed epithelial cancer cells metabolically reprogram adjacent normal fibroblasts.
![Figure 3. HaCaT-fibroblast co-cultures: Cancer-associated fibroblasts show dramatic increases in both ROS production and glucose uptake. (A) ROS-production. (B) Glucose uptake. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 4 days with hTERT-immortalized fibroblasts (RFP[+]). Then, ROS production (a measure of oxidative stress) and glucose uptake (a measure of glycolytic activity) in hTERT-fibroblasts were quantitatively determined by FACS sorting. Note that hTERT-fibroblasts co-cultured with HaCaT-Ras cells show a significant increase in both ROS production (2.5-fold; P = 0.008) and glucose uptake (2.2-fold; P = 0.03). Similarly, hTERT-fibroblasts co-cultured with HaCaT-p65 cells show a significant increase in ROS production (1.9-fold; P = 0.01) and glucose uptake (1.7-fold; P = 0.02). Thus, oncogene-transformed epithelial cancer cells metabolically reprogram adjacent normal fibroblasts.](/cms/asset/fa40b10f-abbe-4c41-841d-6d53e83c9ba7/kccy_a_10925510_f0003.gif)
Figure 4. Cancer-associated fibroblasts Show the largest increases in ROS production and glucose uptake, as directly compared with adjacent epithelial cancer cells. (A) ROS-production. (B) Glucose uptake. Data originally presented in and is presented again in , in another format. In this case, the absolute magnitude of ROS production and glucose uptake in epithelial cancer cells and fibroblasts is directly compared, side-by-side, on the same graphs. This allows one to better appreciate that although the epithelial cancer cells harbor the activated oncogenes, their largest effects on cellular metabolism actually occur in neighboring normal fibroblasts. Thus, the “bystander” effect of oncogenes on the tumor microenvironment is one of the most significant metabolic effects, in terms of metabolic reprogramming.
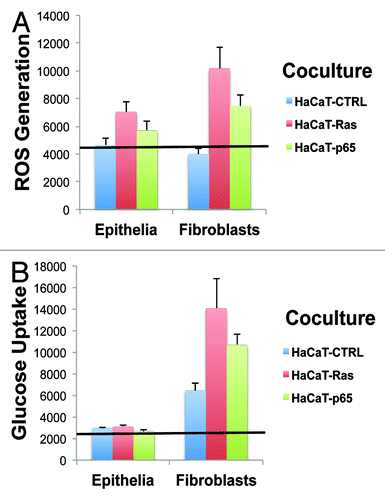
Figure 5. Ras oncogene activation and inflammation drive a loss of Cav-1 expression in adjacent cancer-associated fibroblasts. (A) HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 4 days with hTERT-immortalized fibroblasts (GFP+). Then, the cells were fixed and immunostained with specific antibody probes. Note that a loss of stromal Cav-1 occurs only in fibroblasts co-cultured with HaCaT-Ras and HaCaT-p65 cells. HaCaT control cells did not induce a loss of Cav-1 in stromal fibroblasts. DAPI (blue nuclear staining) is also shown for reference. (B) Image quantitation regarding the oncogene-induced loss of stromal Cav-1 is presented; note that there is a significant 2–3-fold reduction in Cav-1 expression.
![Figure 5. Ras oncogene activation and inflammation drive a loss of Cav-1 expression in adjacent cancer-associated fibroblasts. (A) HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 4 days with hTERT-immortalized fibroblasts (GFP+). Then, the cells were fixed and immunostained with specific antibody probes. Note that a loss of stromal Cav-1 occurs only in fibroblasts co-cultured with HaCaT-Ras and HaCaT-p65 cells. HaCaT control cells did not induce a loss of Cav-1 in stromal fibroblasts. DAPI (blue nuclear staining) is also shown for reference. (B) Image quantitation regarding the oncogene-induced loss of stromal Cav-1 is presented; note that there is a significant 2–3-fold reduction in Cav-1 expression.](/cms/asset/2a3361cc-70f8-4e66-8368-2757b3894b69/kccy_a_10925510_f0005.gif)
Figure 6. Ras oncogene activation and inflammation drive the upregulation of MCT4 expression in adjacent cancer-associated fibroblasts. (A) HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 4 days with hTERT-immortalized fibroblasts (GFP+). Then, the cells were fixed and immunostained with specific antibody probes. Note that MCT4 expression is increased most significantly in fibroblasts co-cultured with HaCaT-Ras and HaCaT-p65 cells. In contrast, HaCaT control cells only induced a mild or modest increase in stromal MCT4 expression. DAPI (blue nuclear staining) is also shown for reference. (B) Image quantitation regarding the oncogene-induced upregulation of stromal MCT4 at day 4 is presented; note that there is a significant 3-fold upregulation of MCT4 expression. P values vs. fibroblasts alone (P < 0.0007) and vs. HaCaT-CTRL co-cultures (P < 0.004) are both shown. (C) Image quantitation regarding the oncogene-induced upregulation of stromal MCT4 at day 5 is presented; note that there is a significant 4-fold upregulation of MCT4 expression. P values vs. fibroblasts alone (P < 0.01) and vs. HaCaT-CTRL co-cultures (P < 0.001) are both shown.
![Figure 6. Ras oncogene activation and inflammation drive the upregulation of MCT4 expression in adjacent cancer-associated fibroblasts. (A) HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 4 days with hTERT-immortalized fibroblasts (GFP+). Then, the cells were fixed and immunostained with specific antibody probes. Note that MCT4 expression is increased most significantly in fibroblasts co-cultured with HaCaT-Ras and HaCaT-p65 cells. In contrast, HaCaT control cells only induced a mild or modest increase in stromal MCT4 expression. DAPI (blue nuclear staining) is also shown for reference. (B) Image quantitation regarding the oncogene-induced upregulation of stromal MCT4 at day 4 is presented; note that there is a significant 3-fold upregulation of MCT4 expression. P values vs. fibroblasts alone (P < 0.0007) and vs. HaCaT-CTRL co-cultures (P < 0.004) are both shown. (C) Image quantitation regarding the oncogene-induced upregulation of stromal MCT4 at day 5 is presented; note that there is a significant 4-fold upregulation of MCT4 expression. P values vs. fibroblasts alone (P < 0.01) and vs. HaCaT-CTRL co-cultures (P < 0.001) are both shown.](/cms/asset/ed40c9e4-8dd6-4054-b227-bb19135a9f60/kccy_a_10925510_f0006.gif)
Figure 7. Oxidative stress mediates the upregulation of MCT4 in cancer-associated fibroblasts: rescue with NAC, a powerful antioxidant. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 5 days with hTERT-immortalized fibroblasts (GFP+). Then, the cells were fixed and immunostained with specific antibody probes. Note that MCT4 expression is increased most significantly in fibroblasts co-cultured with HaCaT-Ras cells and HaCaT-p65 cells, and that this can be reversed or prevented by the addition of the powerful antioxidant, N-acetyl cysteine (NAC) [10 mM], to the culture media. (A) HaCaT control co-cultures; (B) HaCaT-Ras co-cultures; (C) HaCaT-p65 co-cultures. DAPI (blue nuclear staining) is also shown for reference.
![Figure 7. Oxidative stress mediates the upregulation of MCT4 in cancer-associated fibroblasts: rescue with NAC, a powerful antioxidant. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 5 days with hTERT-immortalized fibroblasts (GFP+). Then, the cells were fixed and immunostained with specific antibody probes. Note that MCT4 expression is increased most significantly in fibroblasts co-cultured with HaCaT-Ras cells and HaCaT-p65 cells, and that this can be reversed or prevented by the addition of the powerful antioxidant, N-acetyl cysteine (NAC) [10 mM], to the culture media. (A) HaCaT control co-cultures; (B) HaCaT-Ras co-cultures; (C) HaCaT-p65 co-cultures. DAPI (blue nuclear staining) is also shown for reference.](/cms/asset/999f72ba-f0f7-4915-a959-cb4c98930848/kccy_a_10925510_f0007.gif)
Figure 8. NAC quantitatively reduces oncogene-induced expression of MCT4 in cancer associated fibroblasts. (A) HaCaT-Ras co-cultures; (B) HaCaT-p65 co-cultures. MCT4 expression, illustrated in , was subjected to image quantitation, as detailed under “Materials and Methods”. Note that HaCaT-Ras and HaCaT-p65 cells significantly induce MCT4 expression in adjacent stromal fibroblasts, during co-culture. However, treatment with NAC [10 mM] reduced stromal MCT4 expression levels by nearly 3-fold, approaching baseline levels. Thus, NAC effectively functions as an “MCT4 inhibitor”.
![Figure 8. NAC quantitatively reduces oncogene-induced expression of MCT4 in cancer associated fibroblasts. (A) HaCaT-Ras co-cultures; (B) HaCaT-p65 co-cultures. MCT4 expression, illustrated in Figure 7, was subjected to image quantitation, as detailed under “Materials and Methods”. Note that HaCaT-Ras and HaCaT-p65 cells significantly induce MCT4 expression in adjacent stromal fibroblasts, during co-culture. However, treatment with NAC [10 mM] reduced stromal MCT4 expression levels by nearly 3-fold, approaching baseline levels. Thus, NAC effectively functions as an “MCT4 inhibitor”.](/cms/asset/1c2657c5-e200-4068-84fe-0e8b08a455b0/kccy_a_10925510_f0008.gif)
Figure 9. Co-culture with fibroblasts upregulates the expression of MCT1 in Ras-transformed and NFkB-expressing epithelial cells. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 5 days with hTERT-immortalized fibroblasts (GFP+). Alternatively, HaCaT cells were cultured alone (without fibroblasts) for the same amount of time. Then, the cells were fixed and immunostained with specific antibody probes. Note that MCT1 expression and plasma membrane localization is increased most significantly in HaCaT-Ras cells and HaCaT-p65 cells co-cultured with fibroblasts, relative to the same HaCaT cells cultured alone. Insets at higher magnification are shown to highlight the plasma membrane staining of MCT1. DAPI (blue nuclear staining) is also shown for reference.
![Figure 9. Co-culture with fibroblasts upregulates the expression of MCT1 in Ras-transformed and NFkB-expressing epithelial cells. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were co-cultured for 5 days with hTERT-immortalized fibroblasts (GFP+). Alternatively, HaCaT cells were cultured alone (without fibroblasts) for the same amount of time. Then, the cells were fixed and immunostained with specific antibody probes. Note that MCT1 expression and plasma membrane localization is increased most significantly in HaCaT-Ras cells and HaCaT-p65 cells co-cultured with fibroblasts, relative to the same HaCaT cells cultured alone. Insets at higher magnification are shown to highlight the plasma membrane staining of MCT1. DAPI (blue nuclear staining) is also shown for reference.](/cms/asset/2436d9d3-1bac-4467-994c-d682c7d22526/kccy_a_10925510_f0009.gif)
Figure 10. Homotypic cultures of epithelial cancer cells show that Ras-transformation increases ROS production and MCT4 expression. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were cultured alone and then subjected to either FACS analysis to determine ROS production (A), or immunostaining with antibodies directed against MCT4, a marker of oxidative stress (B). Note that Ras-activation in HaCaT cells significantly increases ROS production (1.5-fold; P = 0.02) and MCT4 expression plasma membrane staining. DAPI (blue nuclear staining) is also shown for reference. (C) shows ROS production in HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) alone or during co-culture with fibroblasts, by comparing data presented in and ; these data are derived from the same experiments. Note that co-culture of normal and epithelial cancer cells with fibroblasts reduced ROS-production in all cases, indicating that fibroblasts may help induce an antioxidant response in adjacent epithelial cells and protect these epithelial cells from oxidative stress.
![Figure 10. Homotypic cultures of epithelial cancer cells show that Ras-transformation increases ROS production and MCT4 expression. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were cultured alone and then subjected to either FACS analysis to determine ROS production (A), or immunostaining with antibodies directed against MCT4, a marker of oxidative stress (B). Note that Ras-activation in HaCaT cells significantly increases ROS production (1.5-fold; P = 0.02) and MCT4 expression plasma membrane staining. DAPI (blue nuclear staining) is also shown for reference. (C) shows ROS production in HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) alone or during co-culture with fibroblasts, by comparing data presented in Figure 2 and Figure 10A; these data are derived from the same experiments. Note that co-culture of normal and epithelial cancer cells with fibroblasts reduced ROS-production in all cases, indicating that fibroblasts may help induce an antioxidant response in adjacent epithelial cells and protect these epithelial cells from oxidative stress.](/cms/asset/067b57f1-8626-4df6-8be9-2a94b7e2847f/kccy_a_10925510_f0010.gif)
Figure 11. Ras oncogene activation and inflammation both drive the upregulation of mitochondrial activity in epithelial cancer cells. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were cultured alone and then subjected to FACS analysis to determine mitochondrial activity, via MitoTracker staining. Note that HaCaT-Ras cells (1.5-fold; P = 0.009) and HaCaT-p65 cells (1.6-fold; P = 0.006) both show a significant increase in MitoTracker activity staining.
![Figure 11. Ras oncogene activation and inflammation both drive the upregulation of mitochondrial activity in epithelial cancer cells. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were cultured alone and then subjected to FACS analysis to determine mitochondrial activity, via MitoTracker staining. Note that HaCaT-Ras cells (1.5-fold; P = 0.009) and HaCaT-p65 cells (1.6-fold; P = 0.006) both show a significant increase in MitoTracker activity staining.](/cms/asset/cfc24ed7-6b84-40c5-9689-b620ba44b58a/kccy_a_10925510_f0011.gif)
Figure 12. Antioxidant treatment selectively inhibits mitochondrial biogenesis in Ras-transformed epithelial cancer cells. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were cultured alone (in the absence or presence of NAC [10 mM]) and then subjected to immunostaining with TOMM20, a marker of mitochondrial mass. Note that HaCaT-Ras cells show the most significant increase in mitochondrial mass, and that this is strictly dependent on oxidative stress. DAPI (blue nuclear staining) is also shown for reference.
![Figure 12. Antioxidant treatment selectively inhibits mitochondrial biogenesis in Ras-transformed epithelial cancer cells. HaCaT epithelial cells (control, H-Ras [G12V], or NFkB [p65]) were cultured alone (in the absence or presence of NAC [10 mM]) and then subjected to immunostaining with TOMM20, a marker of mitochondrial mass. Note that HaCaT-Ras cells show the most significant increase in mitochondrial mass, and that this is strictly dependent on oxidative stress. DAPI (blue nuclear staining) is also shown for reference.](/cms/asset/30b77250-c8d9-4acc-b764-d3d8e52f1532/kccy_a_10925510_f0012.gif)
Figure 13. Fibroblasts protect cancer cells against apoptosis, autophagy, and sensecence: quantitation via FACS. (A) Apoptosis. Cell death was quantified by flow cytometry using propidium iodide and Annexin-V-APC. After a total of 4 days in culture, the cells were collected by centrifugation. Then, the annexin V-APC conjugate and propidium iodide was added. Cells were then analyzed by flow cytometry. Note that fibroblasts (GFP[+]) did not affect cell death in HaCaT control cells, but significantly rescued HaCaT-Ras and HaCaT-p65 cells from apoptosis. However, the most significant fibroblast-mediated reductions in cell death were observed with HaCaT-Ras cells, which showed a 2.4-fold reduction in apoptosis. (B and C) Autophagy and senescence. Cells were treated using the FluoReporter lacZ Flow Cytometry Kit (Molecular probes). Assay results were evaluated by flow-cytometry analysis. Note that HaCaT-Ras cells cultured alone showed very dramatic increases in β-GAL activity, as compared directly with HaCaT control cells. However, β-GAL levels were progressively reduced (by >2-fold) in HaCaT-Ras cells during co-culture with hTERT-immortalized fibroblasts, over a 2–4 d period. Thus, fibroblasts induce autophagy-resistance and reverse senescence in HaCaT-Ras cancer cells. (B) Day 2 co-culture; (C) day 4 co-culture.
![Figure 13. Fibroblasts protect cancer cells against apoptosis, autophagy, and sensecence: quantitation via FACS. (A) Apoptosis. Cell death was quantified by flow cytometry using propidium iodide and Annexin-V-APC. After a total of 4 days in culture, the cells were collected by centrifugation. Then, the annexin V-APC conjugate and propidium iodide was added. Cells were then analyzed by flow cytometry. Note that fibroblasts (GFP[+]) did not affect cell death in HaCaT control cells, but significantly rescued HaCaT-Ras and HaCaT-p65 cells from apoptosis. However, the most significant fibroblast-mediated reductions in cell death were observed with HaCaT-Ras cells, which showed a 2.4-fold reduction in apoptosis. (B and C) Autophagy and senescence. Cells were treated using the FluoReporter lacZ Flow Cytometry Kit (Molecular probes). Assay results were evaluated by flow-cytometry analysis. Note that HaCaT-Ras cells cultured alone showed very dramatic increases in β-GAL activity, as compared directly with HaCaT control cells. However, β-GAL levels were progressively reduced (by >2-fold) in HaCaT-Ras cells during co-culture with hTERT-immortalized fibroblasts, over a 2–4 d period. Thus, fibroblasts induce autophagy-resistance and reverse senescence in HaCaT-Ras cancer cells. (B) Day 2 co-culture; (C) day 4 co-culture.](/cms/asset/70c3a6fc-333a-4ffd-8531-d02a9ef0fe4c/kccy_a_10925510_f0013.gif)
Figure 14. HaCaT epithelial-cancer cell co-cultures: Oncogene-transformed cancer cells can metabolically reprogram normal adjacent epithelial cells. (A) ROS-production. (B) Glucose uptake. HaCaT-Ras (RFP[+]) epithelial cancer cells were co-cultured for 4 days with normal HaCaT control cells. Then, ROS production (a measure of oxidative stress) and glucose uptake (a measure of glycolytic activity) in both cell types were quantitatively determined by FACS sorting. Note that normal HaCaT control cells co-cultured with HaCaT-Ras cells show a significant increase in both ROS production (1.7-fold; P = 0.03) and glucose uptake (1.3-fold; P = 6 × 10−5). However, normal HaCaT control cells did not change ROS production or glucose uptake in HaCaT-Ras cells, indicating that HaCaT-Ras cells exert a significant dominant effect over normal epithelial cells. Thus, oncogene-transformed epithelial cancer cells can metabolically reprogram adjacent normal epithelial cells, to establish “metabolic symbiosis”.
![Figure 14. HaCaT epithelial-cancer cell co-cultures: Oncogene-transformed cancer cells can metabolically reprogram normal adjacent epithelial cells. (A) ROS-production. (B) Glucose uptake. HaCaT-Ras (RFP[+]) epithelial cancer cells were co-cultured for 4 days with normal HaCaT control cells. Then, ROS production (a measure of oxidative stress) and glucose uptake (a measure of glycolytic activity) in both cell types were quantitatively determined by FACS sorting. Note that normal HaCaT control cells co-cultured with HaCaT-Ras cells show a significant increase in both ROS production (1.7-fold; P = 0.03) and glucose uptake (1.3-fold; P = 6 × 10−5). However, normal HaCaT control cells did not change ROS production or glucose uptake in HaCaT-Ras cells, indicating that HaCaT-Ras cells exert a significant dominant effect over normal epithelial cells. Thus, oncogene-transformed epithelial cancer cells can metabolically reprogram adjacent normal epithelial cells, to establish “metabolic symbiosis”.](/cms/asset/0038c60f-dfc7-4afe-bed0-e82dff91a450/kccy_a_10925510_f0014.gif)
Figure 15. Diverse oncogenic stimuli metabolically rewire the microenvironment, via a common mechanism. (A)Oxidative stress and inflammation reprogram the microenvironment. Here, we show that activation of Ras or the innate immune response in epithelial cancer cells are both sufficient to actively induce metabolic reprogramming of the tumor microenvironment. ROS production and/or cytokine release drives oxidative stress in adjacent cancer-associated fibroblasts. Oxidative stress in myofibroblasts then mediates the induction of stromal aerobic glycolysis, i.e., “metabolic symbiosis” or the “reverse Warburg effect”. Thus, diverse epithelial oncogenes (H-Ras [G12V] and NFkB [p65]) induce oxidative stress in the tumor stroma. MCT1 and MCT4 are markers of metabolic symbiosis. (B) Oncogenic stimuli converge on stromal MCT4. HaCaT cells which harbor activated oncogenes gain the ability to glycolytically reprogram the tumor microenvironment, via the induction of oxidative stress in cancer-associated fibroblasts. Thus, diverse oncogenes (Ras and NFkB) act via a common mechanism (oxidative stress), to upregulate MCT4 in the tumor stroma. As MCT4 controls the “energy” supply for cancer cells, this makes MCT4 an attractive target, for new therapeutic interventions aimed at “starving” tumor cells. Similarly, other pro-oncogenic stimuli, such as loss of BRCA1, TGF-β secretion, and ethanol, as well as ROS and hydrogen peroxide production, all converge on MCT4 upregulation in the tumor stromal compartment. In addition, all these pro-oncogenic stimuli (RAS, NFkB, BRCA1 loss, TGF-β, ROS/H2O2, and ethanol) also drive a loss of stromal Cav-1 expression in cancer associated fibroblasts. Thus, Cav-1 and MCT4 are stromal biosensors of the transition to malignancy.
![Figure 15. Diverse oncogenic stimuli metabolically rewire the microenvironment, via a common mechanism. (A)Oxidative stress and inflammation reprogram the microenvironment. Here, we show that activation of Ras or the innate immune response in epithelial cancer cells are both sufficient to actively induce metabolic reprogramming of the tumor microenvironment. ROS production and/or cytokine release drives oxidative stress in adjacent cancer-associated fibroblasts. Oxidative stress in myofibroblasts then mediates the induction of stromal aerobic glycolysis, i.e., “metabolic symbiosis” or the “reverse Warburg effect”. Thus, diverse epithelial oncogenes (H-Ras [G12V] and NFkB [p65]) induce oxidative stress in the tumor stroma. MCT1 and MCT4 are markers of metabolic symbiosis. (B) Oncogenic stimuli converge on stromal MCT4. HaCaT cells which harbor activated oncogenes gain the ability to glycolytically reprogram the tumor microenvironment, via the induction of oxidative stress in cancer-associated fibroblasts. Thus, diverse oncogenes (Ras and NFkB) act via a common mechanism (oxidative stress), to upregulate MCT4 in the tumor stroma. As MCT4 controls the “energy” supply for cancer cells, this makes MCT4 an attractive target, for new therapeutic interventions aimed at “starving” tumor cells. Similarly, other pro-oncogenic stimuli, such as loss of BRCA1, TGF-β secretion, and ethanol, as well as ROS and hydrogen peroxide production, all converge on MCT4 upregulation in the tumor stromal compartment. In addition, all these pro-oncogenic stimuli (RAS, NFkB, BRCA1 loss, TGF-β, ROS/H2O2, and ethanol) also drive a loss of stromal Cav-1 expression in cancer associated fibroblasts. Thus, Cav-1 and MCT4 are stromal biosensors of the transition to malignancy.](/cms/asset/c00d2fb7-e6c4-4d4e-8c47-e8e1c35ac0ef/kccy_a_10925510_f0015.gif)
Figure 16. Understanding why cancer behaves as a wound that does not heal. Oncogenic stress stimulates a common metabolic response to “injury” in the tumor microenvironment. Four steps or events are outlined, leading to “metabolic symbiosis”. This idea is consistent with the hypothesis that cancer behaves as a wound that does not heal.
