Figures & data
Figure 1 Evidence for oxidative stress and mitochondrial dysfunction in Cav-1 (−/−) null mouse tissues: ADMA and Ketones. Note that both 3-hydroxybutyrate (BHBA) and asymmetric dimethyl arginine (ADMA) are increased ∼3–4 fold in Cav-1 (−/−) mammary fat pads. Similar results were obtained with lung tissue harvested from Cav-1 (−/−) mice. Importantly, ADMA is a marker of endothelial dysfunction and oxidative stress; it can also drive oxidative stress, as it functions as an uncoupler of NoS family member, inhibiting the production No and producing superoxide instead. In addition, BHBA is a ketone body known to be a marker of mitochondrial dysfunction. oxidative stress induces mitochrondrial dysfunction and visa versa, driving autophagy and mitophagy.
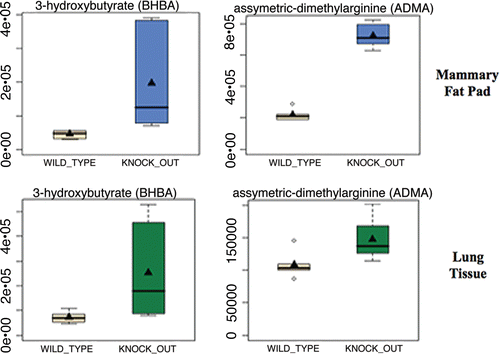
Figure 2 Upregulation of antioxidants in Cav-1 (−/−) mammary fat pads. One compensatory response to oxidative stress is the over-production of anti-oxidants. Note that in Cav-1 (−/−) mammary fat pads an 11-fold increase in Vitamin C (ascorbic acid) and a near three-fold increase in Vitamin E (alpha-tocopherol) were observed.
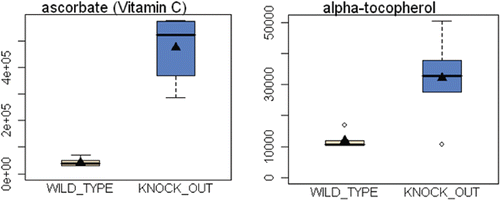
Figure 3 Venn diagrams for the transcriptional overlap between autophagy and tumor stroma from breast cancer patients. Upper panel, overlap with tumor stroma. Note the overlap of 93 genes with a p value of 2.65 × 10−6. Middle panel, Overlap with “recurrence-prone” stroma. Note the overlap of 47 genes with a p value of 2.22 × 10−3. Lower panel, overlap with “metastasis-prone” stroma. Note the overlap of 17 genes with a p value of 5.32 × 10−2.
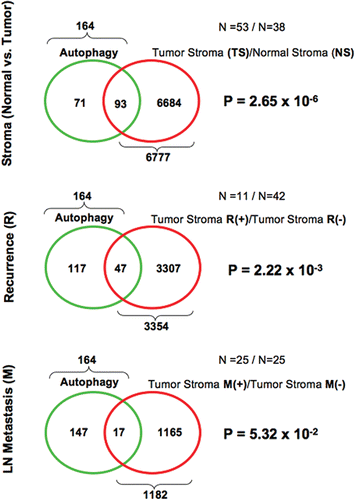
Figure 4 Venn diagrams for the transcriptional overlap between lysosomes and telomere-related genes, with tumor stroma from breast cancer patients. Upper panel, overlap with tumor stroma. Note the overlap of 175 genes with a p value of 1.23 × 10−15. Middle panel, overlap with “recurrence-prone” stroma. Note the overlap of 74 genes with a p value of 2.10 × 10−3. Lower panel, Overlap with “metastasis-prone” stroma. Note the overlap of 38 genes with a p value of 9.67 × 10−5.
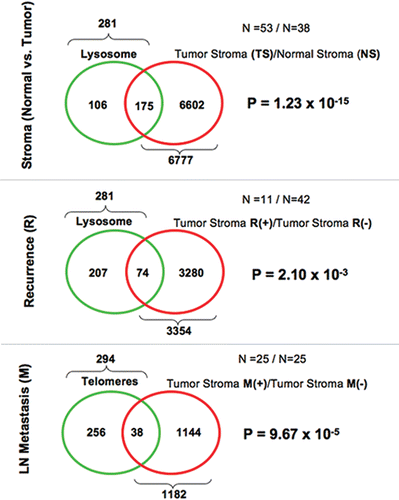
Figure 5 Venn diagrams for the transcriptional overlap between peroxisomes and tumor stroma from breast cancer patients. Upper panel, overlap with tumor stroma. Note the overlap of 204 genes with a p value of 4.25 × 10−12. Lower panel, overlap with “recurrence-prone” stroma. Note the overlap of 101 genes with a p value of 2.76 × 10−5.
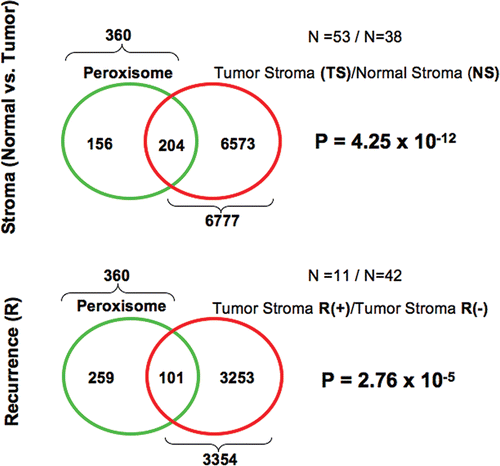
Figure 6 Over-expression of autophagy and mitophagy markers in Cav-1 (−/−) null mammary fat pads: Cathepsin B and BNIP3. To validate the idea that a loss of Cav-1 drives the onset of autophagy, we assessed the expression of established autophagy markers, namely cathepsin B and BNIP3, in Cav-1 (−/−) mammary fat pads. Cathepsin B is a well-known lysosomal protease. BNIP3 is a marker of autophagy that mediates the autophagic destruction of mitochondria. Note that both cathepsin B (the pro-enzyme and activated form) and BNIP3 are significantly overexpressed in Cav-1 (−/−) null mammary fat pads (KO), relative wild-type controls (WT). Immuno-blotting with Cav-1 and ²-actin are shown for comparison. CTSB, cathepsin B.
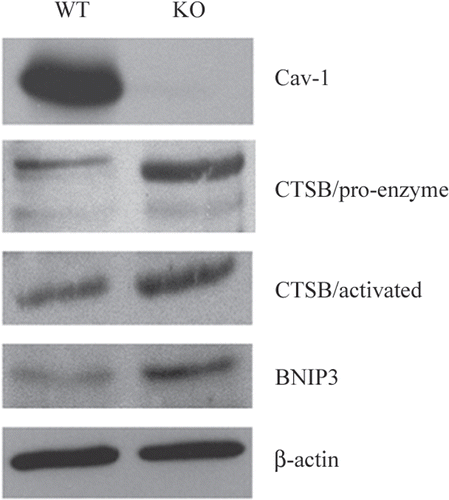
Figure 7 Understanding a lethal tumor micro-environment: Oxidative stress drives stromal autophagy/mitophagy, providing stromal-derived nutrients for epithelial cancer cells. Here, using metabolic, transcriptional mRNA and miR profiling, we have identified that loss of stromal Cav-1 induces oxidative stress, mitochondrial dysfunction and autophagy/mitophagy in the tumor micro-environment. This model would then provide recycled chemical building blocks (nutrients, amino acids, energy-rich metabolites, nucleotides) derived from stromal cells (fibroblasts) that then could be harnessed by epithelial cancer cells to promote tumor growth. Mitochondrial dysfunction and mitophagy would result in aerobic glycolysis in stromal cells, explaining our previous observations on the “reverse Warburg effect”. Many of the key components we have identified here through metabolic and micro-RNA profiling are shown in BOLD: miR-31, miR-34c, ADMA, essential amino acids (AA's), nucleotides, pyruvate, ketones (BHB) and TCA cycle intermediates.
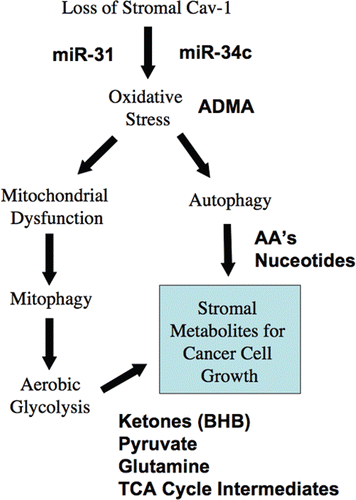
Figure 8 Can ketones fuel tumor growth? Here, we propose that ketones produced in the tumor micro-enviroment (in cancer associated fibroblasts) could fuel the growth of adjacent epithelial cancer cells. Ketone-producing enzymes (in the fibroblasts) and ketone re-utilizing enzymes (in the epithelial cancer cells) are shown in bold. Transfer of ketones would be accomplished by monocarboxylate transporters (MCTs). Normally the same scheme is used by the liver (for ketone production) and the brain (for ketone re-utilization) during extreme fasting or starvation, to maintain neuronal function. Thus, the liver cells are the cancer fibroblasts and the epithelial cells are the neurons. Interestingly, Cav-1 (−/−) stromal cells and the tumor stroma both show a shift towards liver-specific gene and protein expression. For example, Cav-1 (−/−) stromal cells produce alpha-fetoprotein and albumin, as seen by proteomics.Citation15 Alpha-fetoprotein expression has been been previously localized to cancer-associated fibroblasts in human breast cancers.Citation80 The enzymes involved in ketone metabolism are as follows: ACYL, ATP citrate lyase (cytosolic); HMGCS1/2, 3-hydroxy-3-methylglutaryl-Coenzyme A synthase 1 (cytosolic)/2 (mitochondrial); HMGCL, 3-hydroxymethyl-3-methylglutaryl-Coenzyme A lyase; HMGCLL1, 3-hydroxymethyl-3-methylglutaryl-Coenzyme A lyase-like 1; BDH1/2, 3-hydroxybutyrate dehydrogenase, type 1 (mitochondrial)/type 2 (cytosolic); ACAT1/2, acetyl-Co-enzyme A acetyltransferase 1 (mitochondrial)/2 (cytosolic); OXCT1/2, 3-oxoacid CoA transferase 1 (mitochondrial)/2 (testis-specific). We propose that the production of ketone bodies results from Acetyl-CoA derived from pyruvate, via pyruvate dehydrogenase (PDH), and not from the beta-oxidation of fatty acids, because Cav-1 (−/−) null mice have a defect in the beta-oxidation of fatty acids (reviewed in ref. Citation16). this would also mechanistically explain why lactate does not accumulate. Interestingly, ACLY (a cytosolic enzyme) may also contribute to ketone production by converting citrate (a TCA metabolite) to Acetyl-CoA. This also results in the production of oxaloacetate, another TCA metabolite.
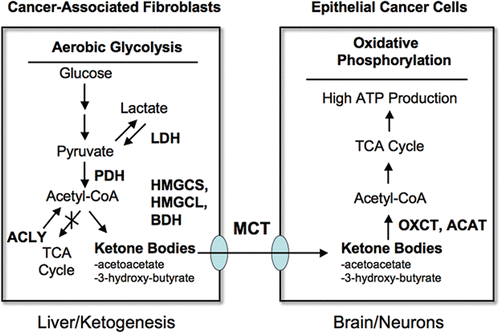
Figure 9 The autophagic tumor stroma model of cancer: Resolving the autophagy paradox in cancer therapy. Here, we propose that autophagy/mitophagy (AM) in the tumor stroma may be sustaining tumor growth. The large black arrow signifies energy transfer (E.T.) from the stromal cancer associated fibroblasts (CAFs) to the epithelial cancer cells, via stromal autophagy/mitophagy. Thus, inhibition of autophagy in the tumor stroma would be expected to halt or reverse tumor growth. This could explain the effectiveness of known autophagy inhibitors as anti-tumor agents,Citation52 such as chloroquine and 3-methyladenine (Upper part). Conversely, induction of autophagy in epithelial cancer cells would also be expected to block or inhibit tumor growth (Lower part). This idea would also explain the anti-tumor activity of agents that activate autophagy, such as mTOR inhibitors.Citation65 Thus, using this model, compounds that either systemically block or activate autophagy would both have the same net effect, which is to disrupt the metabolic coupling between the epithelial cancer cells and the tumor stromal fibroblasts. This model directly resolves the long-lived “autophagy paradox”, that both systemic inhibition of autophagy and systemic stimulation of autophagy have the same net effect, which is to inhibit tumor growth. E.T., energy transfer; AM+, increased autophagy/mitophagy; AM−, decreased autophagy/mitophagy; Rx, therapy with autophagy promoters or inhibitors.
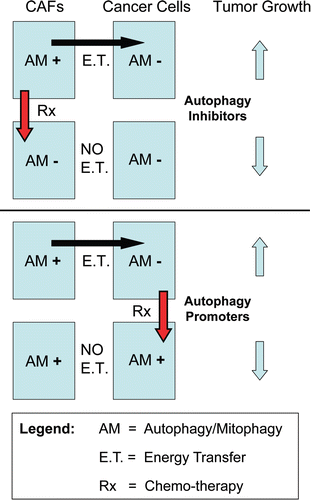
Table 1 Metabolomic analysis of mammary fat pads from Cav-1 (−/−) deficient mice
Table 2 Metabolomic analysis of mammary fat pads and lung tissue from Cav-1 (−/−) deficient mice
Table 3 Upregulation of autophagy/mitophagy related gene transcripts in Cav-1 (−/−) stromal cells
Table 4 Upregulation of gene transcripts encoding lysosomal proteins in Cav-1 (−/−) stromal cells
Table 5 Upregulation of telomerase and selected redox-related gene transcripts in Cav-1 (−/−) stromal cells
Table 6 Upregulation of autophagy/mitophagy related gene transcripts in the tumor stroma from human breast cancer patients
Table 7 Upregulation of gene transcripts encoding lysosomal proteins in the tumor stroma from human breast cancer patients
Table 8 Upregulation of telomerase and selected redox-related gene transcripts in the tumor stroma from human breast cancer patients
Table 9 Transcriptional profiling of human breast cancer tumor stroma: ADMA and BHB metabolism
Table 10 Upregulation of miR's in Cav-1 (−/−) null stromal cells