Figures & data
Figure 1 p27Kip1 is mislocalized in Shh-mediated medulloblastoma. (A) In cerebellar development of post-natal day 7, the peak for CGNP proliferation, p27Kip1 (green) is expressed in post-mitotic region, external granule layer b (EGLb), molecular layer (ML) and differentiated region inner granule layer (IGL), whereas aberrant Shh signaling leads to p27Kip1 mislocalization and increased CGNP proliferation throughout the EGL. Proliferation marker PCNA (red) stains CGNPs in proliferating region EGLa. (B) Wild-type and SmoA1 CGNPs were plated, treated with Shh and measured for proliferation by quantification of phospho-Histone H3. (C) While non-tumor cells undergo cell cycle exit and/or differentiation via p27Kip1 nuclear localization in the IGL, the Shh-induced medulloblastoma maintains misregulated phosphorylation of p27Kip1 at Ser10 and Thr187, which leads to mislocalization and inhibition of cell cycle regulation. (D) Non-tumors in NeuroD2-SmoA1 mice have nuclear p27Kip1 (first column), whereas SmoA1 medulloblastoma have cytoplasmic p27Kip1 (second column) and high proliferation (third and fourth column).
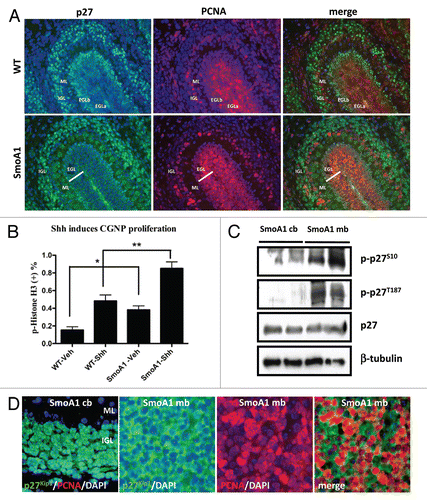
Figure 2 (A–C) p27Kip1 loss accelerates medulloblastoma incidence in SmoA1 mice. (A) p27Kip1 loss accelerates medulloblastoma incidence in homozygous and hemizygous mice for SmoA1 transgene. (B and C) SmoA1 mice heterozygous for p27Kip1 have a dramatic decreased survival latency compared to homozygous and nullizygous for p27Kip1.
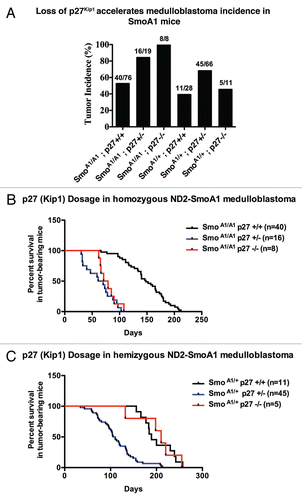
Figure 2D p27Kip1 loss accelerates medulloblastoma incidence in SmoA1 mice. (D) Western blot analysis of cell cycle indicators in NeuroD2-SmoA1 medulloblastomas arising in wild-type, p27+/− and p27−/− mice.
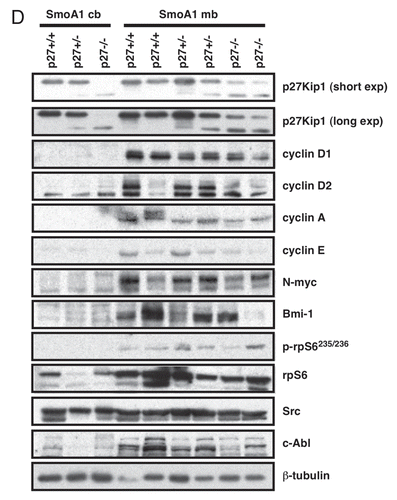
Figure 3 Cyclin D/Cdk4/6 assembly is dependent on p27Kip1. (A) Immunoprecipitation shows p27Kip1 interacts with cyclin D1, Cdk4/6 in SmoA1 medulloblastoma. (B) Immunoprecipitation shows decreased cyclin D1/Cdk6 interaction as a result of p27Kip1 loss. Tumor proliferation benefits as cyclin E/p27Kip1 is decreased, suggesting p27Kip1 is haploinsufficient as a tumor suppressor.
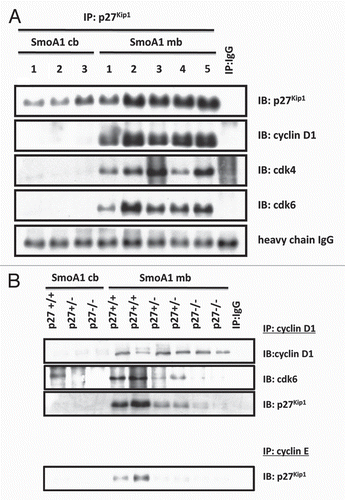
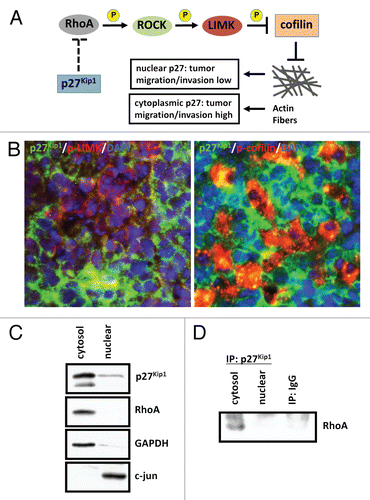
Figure 4 p27Kip1 plays a role in cell motility in SmoA1 medulloblastoma. (A) Schematic diagram of p27Kip1 regulating cell motility in tumor cells. (B) Immunofluorescence shows p27Kip1 is inversely correlated with stabilized cell motility, as evidenced by p27Kip1 (green), active phosphorylated-LIMK to prevent cell motility (red, left panel) and inactive phosphorylation of LIMK substrate and actin-depolymerization protein cofilin (red, right panel) in SmoA1 medulloblastoma. (C) Subcellular fractionation of SmoA1 medulloblastoma shows p27Kip1 and cell motility regulator RhoA are localized mainly in the cytoplasm. GAPDH serves as cytosolic control and c-jun as nuclear control. (D) Immunoprecipitation of p27Kip1 reveals interaction with RhoA. This data suggests p27Kip1 is needed to positively regulate cell motility in SmoA1 medulloblastoma.