Figures & data
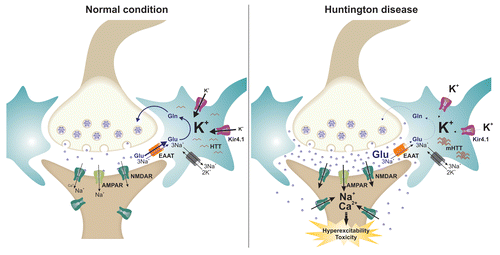
Figure 1. A putative model linking astrocytal Kir4.1 channel and glutamate homeostasis in Huntington disease. In normal condition, glutamate released in the synaptic cleft is rapidly removed by astroctal EAAT transporters that serve o terminate the excitatory signal and preventing neuronal excitotoxicity. Under pathological condition such as Huntington disease, decreased Kir4.1 channel activity leads to aberrant K+ homeostasis and transmembrane K+ gradient disturbing EAAT activity and astrocytal glutamate uptake. Accumulation of glutamate in the synaptic cleft causes in turn neuronal hyperexcitability and in the long-term cellular toxicity, possibly by activation of extrasynaptic NMDA receptors.