Figures & data
Figure 1. Methylated DNA Immunoprecipitation (MeDIP) protocol. The black arrow indicates the introduction of exogenous controls described in this study.
Table 1. Primers sequences for Spike Positive and Negative Controls
Figure 2. Digestion patterns of positive and negative controls with the restriction enzymes AciI, MspI and HpaII on 2% Metaphor agarose gel. Lanes 1 and 5 = AciI digestions of positive and negative control, respectively; lane 2 and 6 = MspI digestions of positive and negative control; lanes 3 and 7 = HpaII digestions of positive and negative control; lanes 4 and 8 = undigested positive and negative control; lane 9 = 100 bp DNA Ladder. Image in the bottom row shows virtual gel with the corresponding digestions patterns.
Figure 3. Panel A and C: descriptive statistics and regression analysis for fold enrichments positive control/negative control pre-WGA and post-WGA. Box plots in Panel A show the percentiles and the median of column data. Box outlines define the 25th and 75th percentiles, with a line at the median and whiskers indicate the 10th and 90th percentiles. Panel C shows linear regression for pairs of pre-WGA and post-WGA enrichments. n = 33. Panel B: WGA amplification bias. Black bars represent the amplification rate post-WGA/pre-WGA of different length Lambda DNA fragments spiked in mouse genomic DNA. Error bars show SEM, n = 3.
Figure 4. Gene specific enrichments after MeDIP followed by WGA in MRC5 fibroblast DNA and in various mouse tissues. (A) Enrichment of the MRC5 internal genes H19 (black bar) and HIST1H2BA (gray bar), both normalized to GAPDH. (B) positive/negative control Enrichment Post-WGA. Error bars show standard error of the means, n = 3 technical replicates. Gene specific enrichments for H19 (C) and Hist1h2ba (D) in mice tissues normalized to either Actb (black bars) or Gapdh (gray bars). Data are mean ± SD; n, 3 biological replicates; AL, ad libitum fed; DR, dietary restricted.
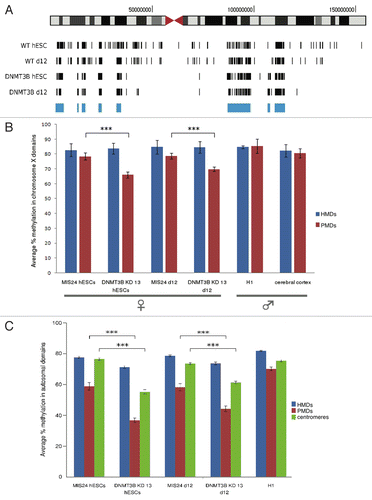
Figure 5. Panels A and B: normalization of endogenous gene enrichment by the negative control reduced experimental variability. A) Average fold enrichments for Hist1h2ba normalized with the endogenous region Actb (left column) or with the spiked negative control (right column) in old mice heart DNA. Data are mean ± SD, n = 3 biological replicates. Means are not significantly different (p = 0.41, Student’s t-test). B) Black bars show Coefficients of Variation (CV) related to estimates in panel A. Panels C and D: normalization of endogenous gene enrichment by the negative control reduced the impact of tissue-specific methylation of the normalizer gene. Panel C shows the comparison between gene specific enrichments for Hist1h2ba calculated with the endogenous normalizer Actb and with the negative control for young colon samples (white bars) and young heart samples (dashed bars). Panel D shows positive control/negative control enrichment post-WGA. Data are mean ± SD, n = 3 biological replicates. * p = 0.04, Student’s t-test. Panels E, F and G: validation of Panel C results by BM sequencing. Panel E shows direct BM sequencing of murine Hist1h2ba; data are mean ± SD, n = 3 biological replicates. Panel F and G show aggregate representation of methylation data obtained by Bisulfite Modified (BM) cloning and sequencing for one colon and one heart sample, respectively. The bars indicate the percentage of clones that showed unmethylated (blue) or methylated (yellow) cytosines at the CpG sites analyzed (adapted from BiQ Analyzer v2.00).
Figure 6. Methylation quantification by direct bisulfite modified DNA sequencing of human MRC5 H19, HIST1H2BA and GAPDH regions (see Table S3 for primers and BM DNA sequences). Panels A, C and E Y axes present data measured for 3 independent technical replicates. Panels B and D present average methylation ± SD across the CpG sites analyzed.
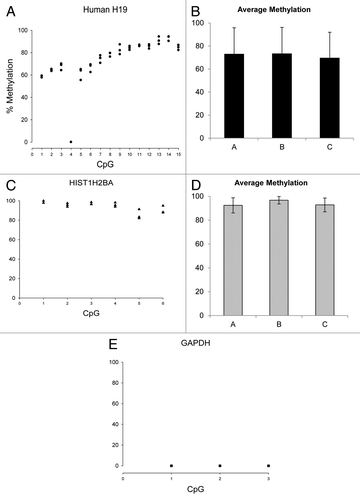
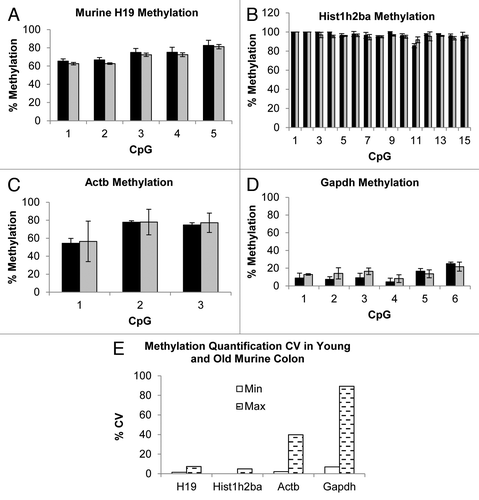
Figure 7. Methylation quantification by direct bisulfite modified DNA sequencing of H19, Hist1h2ba, Actb and Gapdh regions of murine Young (black bars) and Old (gray bars) colon samples (Panels A to D, see Table S3 for primers and BM DNA sequences). Error bars present SD calculated on each single CpGs measurements of 3 biological replicates. Panel E shows minimum (white bars) and maximum (dashed bars) percentage CV determined on all the investigated CpGs measurements.