Figures & data
Figure 1. Comparison of DNA methylation in tumor and normal tissue. (A) A representative screen shot of the normalized DNA methylation data uploaded in the UCSC genome browser. 14T indicates a tumor sample; 14N indicates the matched normal sample. In total, 5 sample pairs are shown. The read-out of MethylCap-seq is semi-quantitative; the number of sequence reads homologous to a genomic region gives a measure for DNA methylation at that region. In the screenshot several regions show DNA methylation. Most peaks of methylation are present in all samples (indicated with gray boxes at the top), except for the methylation at the promoter of LHX5 (red box), which is only present in the tumors and not in the normal tissue. (B) Scatterplot of the median methylation level of normal tissue and tumor. (C) Scatterplot of the standard deviation of normal tissue and tumor. (D) The first two principal components resulting from PCA of all DNA methylation profiles separate the tumors and normal colon samples. The red dots are tumors; the blue dots normal samples.
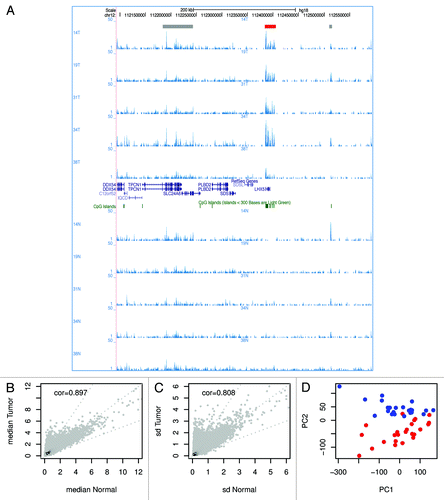
Figure 2. Differentially methylated regions. (A) Pie chart of the 32,9613 analyzed regions. For 9% of the regions hypermethylation and for 14% hypomethylation was observed in at least one patient; for 4% both hypo- and hyper-methylation was detected. (B) Bar graph of the frequency of detection of DMRs. We defined the “support” as the number of sample pairs in which a region was differentially methylated. (C) Hierarchical clustering was performed using the 3,155 high-confidence DMRs and a heatmap of the DNA methylation was made. Columns represent the individual samples; rows the DMRs. Red indicates high methylation; yellow low. Above the columns tumors are marked in red and normal samples in blue. (D) Stacked bar charts showing the distribution of the 329,613 analyzed regions over six genomic elements. The regions are binned by support. Note: the number of differentially methylated regions reduces with the level of support (). Only two hypermethylated regions were recurrent in 23 tumors; one hypomethylated region was recurrent in 10 tumors, and another region in 13 tumors.
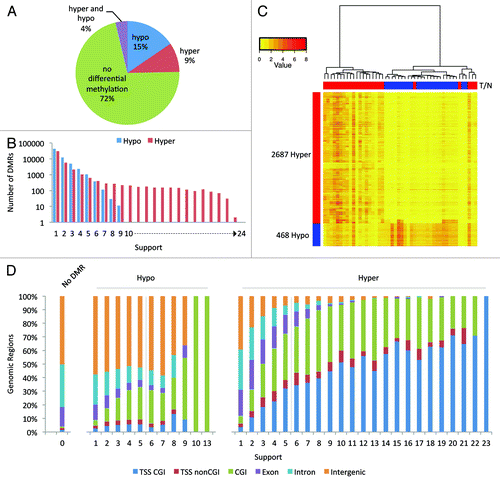
Table 1. Top 10 of high-confidence DMRs. DMRs were sorted on support for hypermethylation and fold difference in expression between HCT116 WT and DKO cells
Figure 3. DMRs with the highest support. Two regions are hypermethylated in 23 of the 24 tumors. (A) Screenshots show that the regions overlap with the promoters of the genes TLX1, TLX1NB, and SDC2. (B) Strip charts with the MethylCap-seq read densities of all samples. The dotted line indicates the mean read density. (C) Bar charts for the RNA-seq results for HCT116 WT and DKO cells. For SDC2 the expression is 25× higher in the DKO compared with the WT; for TLX1 5×; for TLX1NB no sequence reads were obtained in WT and only few in the DKO.
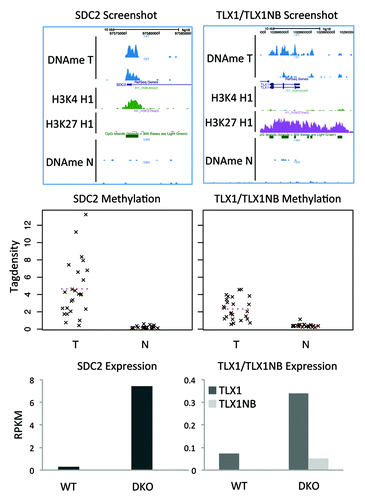
Figure 4. Correlation between HCT116 epigenetic marks and transcripts. (A) Color profiles of MethylCap-seq, H3K4me3, H3K27me3 and CTCF ChIP-seq and DNaseI hypersensitivity sequencing read densities from HCT116. Each profile shows the 10kb regions surrounding the high-confidence hypermethylated DMRs. The DMRs were sorted based on their support. The bar on the left shows the support (high support is red; low support is yellow). Average profiles are shown on top of the color profiles. (B) Density plot of the ratio of the RNA-seq data of HCT116 WT and DKO. The ratios of the RPKM values for DKO over WT are plotted for high-confidence (support hypermethylation ≥ 6; support hypomethylation ≤ 1), low-confidence (support hypermethylation < 6; support hypomethylation ≤ 1), and no DMRs (support hyper- and hypomerhylation = 0).
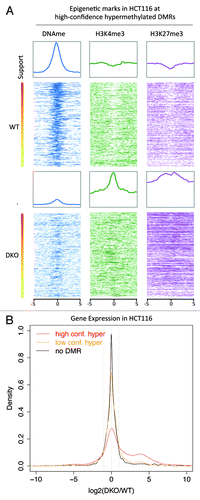
Figure 5. Overlap with bivalent regions in hESC. (A) Color profiles of H3K4me3, H3K27me3, H3K36me3, PolII, and CTCF ChIP-seq and DNaseI hypersensitivity sequencing read densities from hESC H1 (ENCODE). The DMRs are divided into regions overlapping with CGIs with TSS, regions overlapping with CGIs without TSS/Orphan CGIs, and regions not overlapping with CGIs. The regions were sorted on H3K27me3 density. (B) Pie chart showing the overlap of the high-confidence hypermethylated DMRs with H3K4me3 and H3K27me3 peaks from H1. For comparison a set of random regions obtained from the total set of peaks analyzed is included.
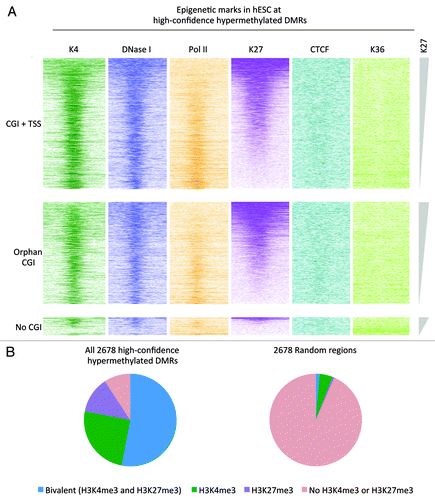
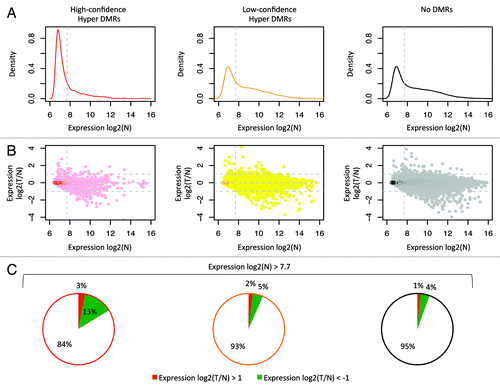
Figure 6. Correlation with gene expression. Analysis of the publically available gene expression data from 26 CRC and matched normal colon samples (GSE25070). On the left data for the high-confidence hyper DMRs; in the middle low-confidence hyper DMRs; on the right regions without differential methylation. (A) Density plots of the mean expression level in normal samples. Dotted line indicates the median expression level calculated from all probes associated with a MethylCap-seq peak. These plots show that a large fraction of the high-confidence hypermethylated DMRs is lowly expressed in normal colon. (B) Scatter plots of the ratio of the mean expression of tumors and normal. Only probes that were expressed above median level in normal showed a more than 2-fold reduction in expression in tumor compared with normal. (C) Pie charts for probes with normal expression above median level. 13.3% of the selected probes that associated with the high-confidence hyper DMRs shows more than 2-fold lower mean expression in tumor compared with normal, whereas this is 4.7% and 4.2% for low-confidence hyper DMRs and peaks without differential methylation, respectively.