Figures & data
Figure 1. Validation of probe β values using bisulfite clone sequencing of FOXL1 and OVOL1. Single colony sequencing of FOXL1 and OVOL1, which were previously identified to be methylated in ccRCC and determined to be methylated with a β value > 0.5, was conducted in some of our samples. Hypermethylated loci (β value > 0.5) also showed a high methylation index (MI > 50%), while low β value loci correlated with a low MI. Bisulfite sequencing rows represent individual alleles, with each circle indicating location and methylation state of a CpG locus (black circle: methylated; white circle: unmethylated). Arrowheads indicate the HumanMethylation450 array locus under investigation. Also listed next to the gene name are the Target IDs.
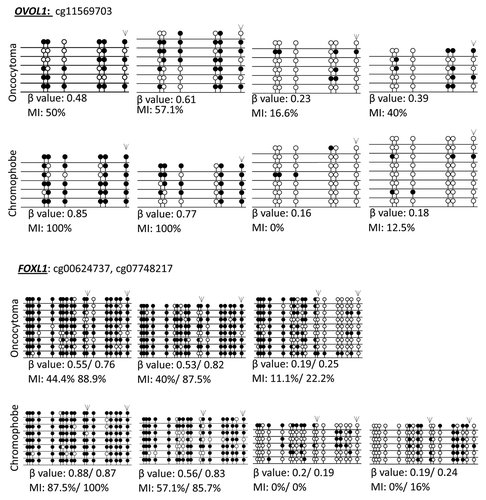
Figure 2. Supervised hierarchical clustering using Euclidean distance complete linkage of the 500 most variable cancer-specific hypermethylated (A) and hypomethylated (B) loci. Below the hierarchical cluster, the top row shows black squares for chromophobe samples and white squares for oncocytoma samples. Gender is denoted in the middle row: female by black squares and male by white squares. Patient age range is indicated in the lower row: 30–49 y, white squares; 50–69 y, gray squares; 70–89 y, black squares. Samples lacking these data are indicated by crossed out boxes. No clustering was observed in relation to gender or age.
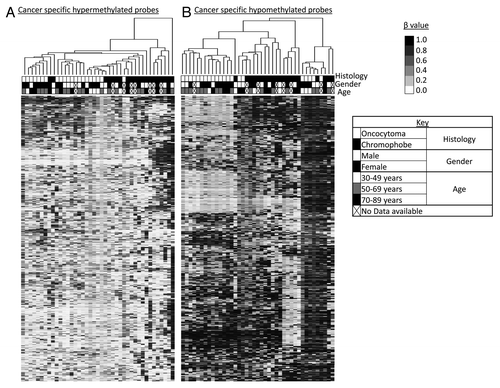
Figure 3. Methylation profiles of chromophobe RCC and renal oncocytoma samples. (A) Global methylation profile was mapped in relation to the 4 normal kidney samples included on the array. Differentially methylated loci were deemed to be all cancer-specific loci showing hyper- or hypo-methylation (1.2% or 4,439 loci for chromophobe samples and 0.6% or 2,383 loci for oncocytoma samples). All other loci not fulfilling this criterion where deemed to be equally methylated to the normal. (B) Methylation profile of cancer-specific loci identified as either hypermethylated (β value > 0.5) or hypomethylated (β value < 0.25) in > 30% of chromophobe RCC and > 30% renal oncocytoma samples. The majority of loci showed hypomethylation in both histologies with less than 10% of cancer-specific probes being hypermethylated. (C) Genomic distribution of cancer-specific hyper- and hypo-methylated CpG loci in relation to their location within known genes. The promoter region indicates loci residing within the 1st exon, 5′UTR, TSS200 and TSS1500 of known genes. CpG distribution did not differ between the two histologies, or between the profiles of hypermethylated and hypomethylated loci. (D) Genomic distribution of cancer-specific hyper- and hypo-methylated CpG loci in relation to CpG density. The majority of hypermethylated loci are shown to reside in areas of high CpG density (CpG islands, north and south shores and north and south shelves): 79.1% for chromophobe samples and 76.0% for oncocytoma samples. Cancer-specific hypomethylated loci are mostly located in isolated/low-density CpG regions known as open sea (65.7% for chromophobe samples and 67.3% for oncocytoma samples).
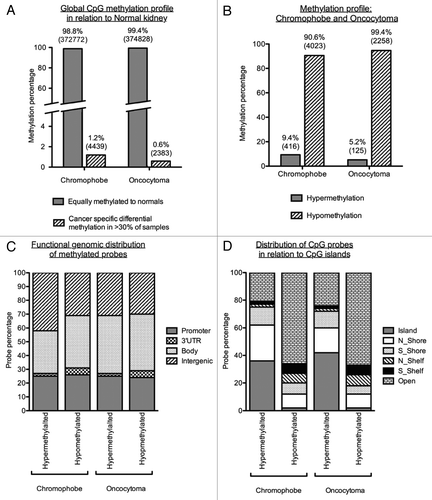
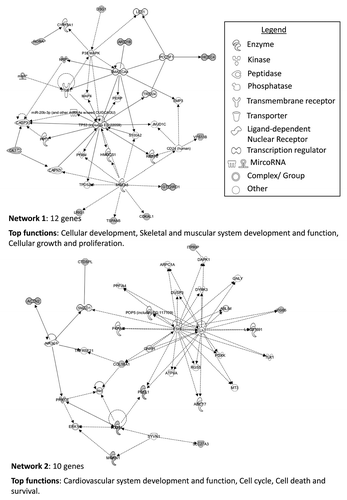
Figure 4. Top three networks identified by Ingenuity associated functional network analysis of genes hypermethylated in > 30% of chromophobe RCC samples. Methylated genes are shown in gray and connecting genes in white. Solid arrows represent direct interaction, dashed arrows highlight indirect interactions, solid joining lines identify protein binding only. IPA analysis identified several key networks with the top three showing interactions and involvement in embryonic development, tissue development and morphology, cell signaling, molecular transport, vitamin and mineral metabolism, cell death and survival, cell cycle and cancer.
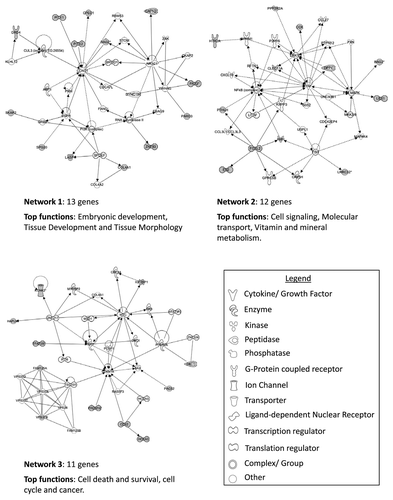
Figure 5. Top three networks identified by Ingenuity associated functional network analysis of genes hypermethylated in > 30% of renal oncocytoma samples. Methylated genes are shown in gray and connecting genes are in white. Solid arrows represent direct interaction, dashed arrows highlight indirect interactions, solid joining lines identify protein binding only. IPA analysis identified networks with involvement in cell signaling and molecular transport, cell morphology, cellular assembly and organization, cellular function and maintenance, cancer, cellular growth and proliferation, and renal and urological disease.
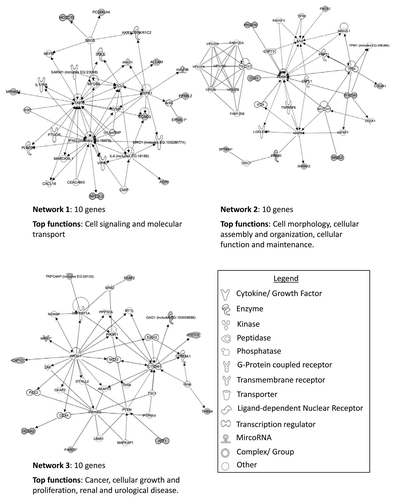
Table 1. Differentially hypermethylated genes between chromophobe RCC and renal oncocytoma samples
Figure 6. Validation of loci β values using bisulfite clone sequencing of NPHP4 and SPG20 genes. (A) Schematic overview of the NPHP4 gene (black boxes represent exons, black lines represent introns and gray boxes represent CpG islands). Black line below CpG island represents region interrogated by primers for bisulfite sequencing including differentially methylated CpG locus (black dash). The differentially methylated CpG resides in a CpG island within 200 bp of the TSS of the gene. (B) Schematic overview of the SPG20 gene. The differentially methylated CpG resides in a north shore in the 5′UTR region of the gene. Bisulfite sequencing rows represent individual alleles, with each circle indicating location and methylation state of a CpG locus (black circle: methylated; white circle: unmethylated). Black arrowheads point to the CpG loci identified on the 450K array as showing significant differential methylation. Bisulfite sequencing of chromophobe RCC and renal oncocytoma samples confirm that the CpG loci in NPHP4 and SPG20 are hypermethylated in chromophobe RCC samples and unmethylated in renal oncocytoma samples.
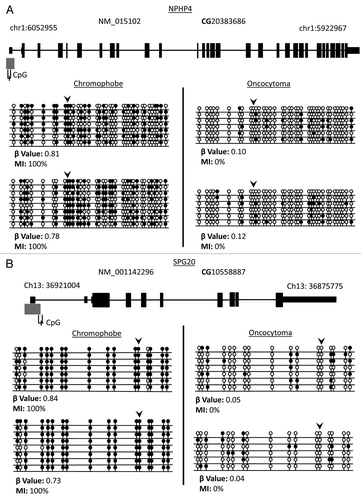
Figure 7. IPA network analysis of genes showing significant differential methylation between chromophobe RCC and renal oncocytoma samples. Top two networks and the interactions between methylated genes (in gray) and connecting genes (in white). Solid arrows represent direct interactions, dashed arrows highlight indirect interactions and solid joining lines identify protein binding.
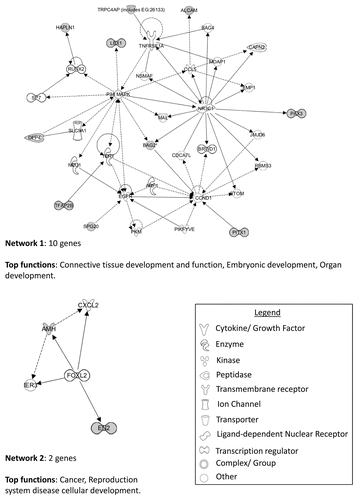
Figure 8. Identification of histologic-specific differential methylation. Genes identified as differentially methylated between oncocytoma and chromophobe samples were compared with the β values for ccRCC and papillary RCC from the TCGA data using one-way ANOVA with Games-Howell post hoc test. (A) Three genes were identified as significantly methylated in chromophobe RCC samples compared with the other histologies. Box plots with whiskers display the median ± maximum and minimum values for each histology. (B) Differentially methylated genes in renal oncocytoma identified two genes. One of them, HOXA9, identified as significantly hypomethylated compared with the other histologies. p values: * > 0.05, ** > 0.01, *** > 0.001
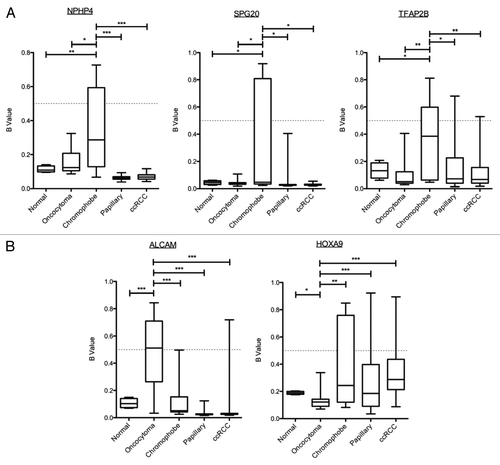
Table 2. Differentially hypomethylated genes between chromophobe RCC and renal oncocytoma samples
Figure 9. IPA network analysis of genes showing significant differential hypomethylation between chromophobe RCC and renal oncocytoma samples. Top two networks and the interactions between hypomethylated genes (gray) and connecting genes (white). Solid arrows represent direct interaction, dashed arrows highlight indirect interactions and solid joining lines identify protein binding. Associated functions include cardiovascular system development and function, cell cycle, cell death and survival, cellular development, skeletal and muscular system development and function, and cellular growth and proliferation.