Figures & data
Figure 1. The workflow to compare pipelines. Six pipelines were considered in this study, as explained in the main text. Using data set A, we run all the pipelines, selecting the one to be tested with data set B, which also allows the evaluation of the correction for batch effect.
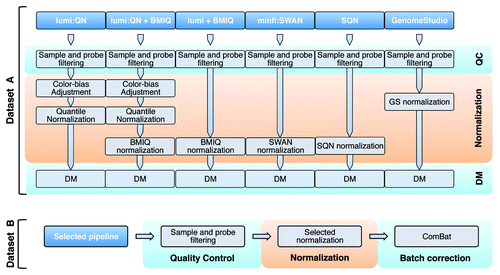
Figure 2. Evaluation of the lumi pipeline. (A) The difference in sample correlation (upper panel) or Euclidean distance (lower panel) between quantile normalization (QN) and raw data. For convenience, only samples represented by two or three technical replicates are shown. A gray bar indicates the samples from monocytes, whereas a black bar denotes the samples from peripheral blood (PB). Replicates belonging to the same pair are consecutively located on each row or column. The color code indicates a decrease (blue) or an increase (yellow) in correlation or distance. (B) The absolute deviation between technical replicates in raw, color adjusted and QN data shows the consistent reduction of the technical variability after normalization. (C and D) The logarithmic ratio between the variability after QN and the variability on raw data are shown for PB (C) and monocytes (D). For each probe, we calculated the average M-values and the corresponding mean absolute difference between technical replicates. The log2 ratio between QN and raw data was used to check the performance of the normalization in reducing the variability and the presence of possible bias for sites with low or high levels of methylation. The red line indicates the loess fitting.
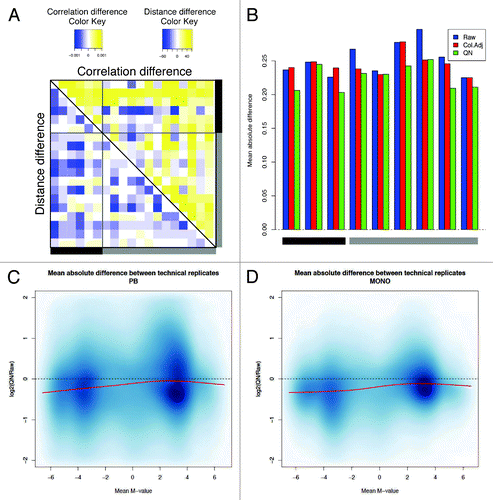
Figure 5. Minfi pipeline incorporate an adjustment for probe design type, which is obtained with SWAN. The raw (A), and SWAN (B) densities of M-values are shown either for each sample, or as the average density for Infinium I or Infinium II probe design.
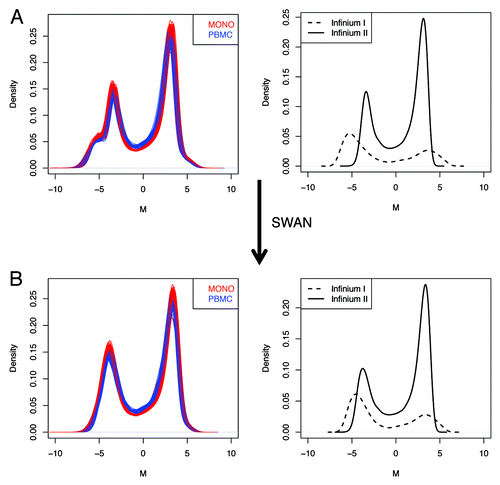
Figure 3. Lumi pipeline does not incorporate an adjustment for probe design type, which is obtained with BMIQ. The raw (A), QN normalized (B) and QN + BMIQ (C) densities of M-values are shown either for each sample, or as the average density for Infinium I or Infinium II probe design. (D) BMIQ alone is also suitable for eliminating the probe design type bias.
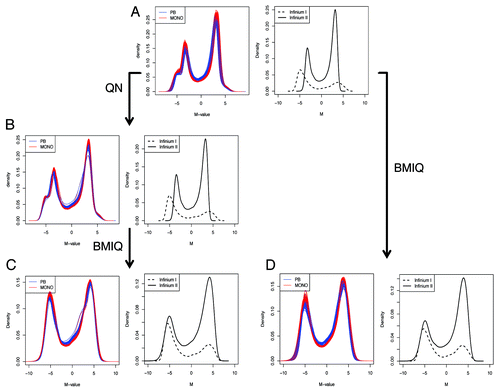
Figure 6. Elimination of probe design bias using BMIQ. (A) The absolute deviation between technical replicates after QN, QN + BMIQ or BMIQ show the consistent reduction of the technical variability after adjusting for probe design type. To calculate the difference, β-values were considered in this case. (B) The mean absolute difference between probe pairs (as defined in method section) shows that the reduction of the technical noise due to different design type, is optimally obtained using BMIQ, which is superior to SWAN, SQN or GS.
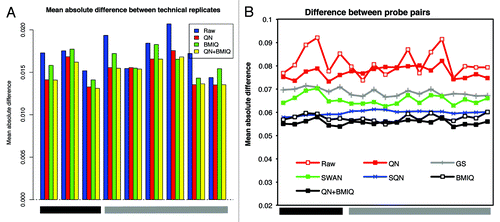
Figure 4. Evaluation of the minfi pipeline. (A) The difference in sample correlation (upper panel) or Euclidean distance (lower panel) between SWAN and raw data. For convenience, only samples represented by two or three technical replicates are shown. Color codes and positions of the samples are the same as . (B) The absolute deviation between technical replicates in raw and SWAN-normalized data shows the reduction of the technical variability after normalization. (C and D) The logarithmic ratio between the variability after SWAN and the variability on raw data are shown for PB (C) and monocytes (D). For each probe, we calculated the average M-values and the corresponding mean absolute difference between technical replicates, as explained in .
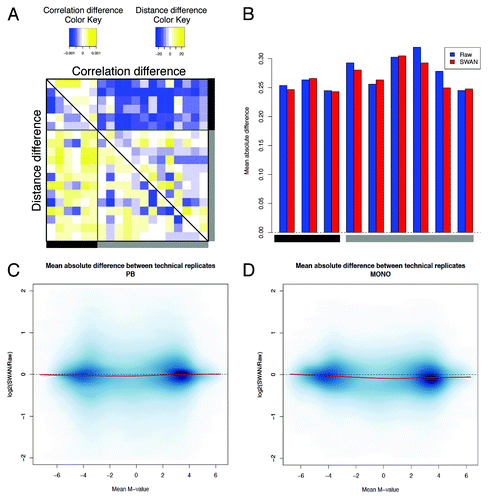
Figure 7. The analysis of DM sites is influenced by the normalization process. (A) Volcano plots show the p value vs. the difference in methylation as calculated by limma. Comparable amplitude in methylation difference is obtained only after adjusting with QN + BMIQ, BMIQ but not with QN only. (B) Spearman correlation between P-values obtained after different normalization options. The correlation was calculated progressively including probes from the ranked list of CpG sites, as described in Methods section. (C) Venn diagram showing the number of DM sites obtained after different pipelines. The threshold for claiming DM was FDR Adjusted p < 0.05 AND absolute(ΔM) > 1. (D) PCA of all probes denotes the pattern of variability. The color indicates the smoothed density (black = low, yellow = high). The first PC accounts for the average methylation level, while the second PC indicates the direction of the methylation change in monocytes as compared with PB. DM sites are indicated as dots, with different colors for increased (green) or decreased (white) methylation. There is no sign of different behavior of type I or type II probes.
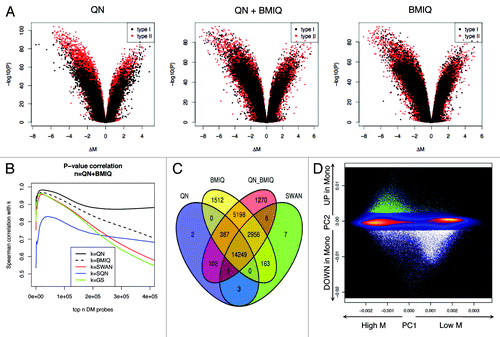
Table 1. Number of differentially methylated CpG sites resulting from the indicated pipelines (columns) and thresholds (rows)
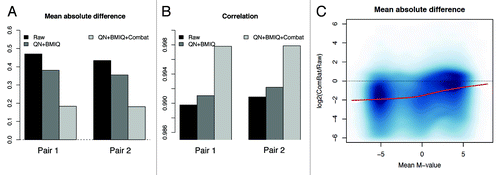
Figure 8. The elimination of unwanted batch effect further reduces the technical variability. After correcting for batch effect, we observed an increase of correlation (A) and a decrease of the absolute deviation (B) between technical replicates. (C) The logarithmic ratio between the variability after QN + BMIQ + ComBat and the variability on raw data are shown, calculated as explained in .