Figures & data
Table 1. Distribution of differentially methylated regions in brain and blood DNA from Parkinson disease patients
Figure 1. DNA methylation profiles of brain and blood in Parkinson disease cases. (A) Comparison of the fractions of differentially methylated loci that showed gain or loss of methylation in PD brain and blood. (B) Distribution of average β values across samples and tissues showing enrichment in low-methylation (LMF, β values < 20%) and high-methylation fractions (HMF, β values > 80%). IMF, intermediate methylated fraction (β values < 20% and > 80%). *Significant difference in PD blood in comparison to control subjects’ blood (p < 0.05) and to PD brains (p < 0.001, One-way ANOVA with correction for multiple observations and Bonferroni post-hoc test). (C) CpG neighborhood context analysis in brain and blood samples that showed increased methylation (IM) or decreased methylation (DM) in PD. CG islands Shores are defined as regions up to 2 Kb from the CGi Start or End; Shelves are defined as the next 2 Kb boundaries from CGi shores. (D) Genomic location distribution of differentially methylated loci in PD brain and blood samples. TSS1500, CpG sites within 200–1500 bp from the transcription-starting site (TSS). TSS200, CpG sites within 200 bp from the TSS. Body, gene body regions. (E) Gene ontology analysis of annotated genes showing differential methylation clustering by biological process.

Figure 2. Correlation of β-values between brain and blood in PD samples. Linear regression and Spearman correlation coefficients were calculated for the methylation values (β) of individual PD samples for the top 20 DM probes that showed increased (A) or decreased (B) methylation in only one tissue or that co-varied in both brain and blood. (C) Fisher’s exact probability test on DM probes testing the null hypothesis that the observed changes in methylation in brain were independent of the changes on the same probes in blood.
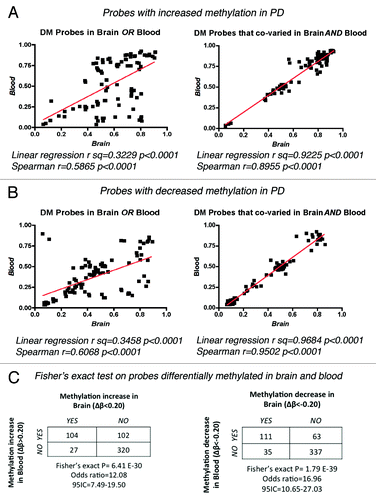
Table 2. Differentially methylated autosomal genes that co-varied in brain and blood DNA from PD patients
Figure 3. Effects of drug regimen, age and disease duration on DNA methylation. (A) Box plot showing the average β-value of the total probes interrogated per individual PD case. Age at time of death is indicated in (black). Duration of disease, calculated as years elapsed since first PD diagnosis until time of death, is indicated in red. Drug treatment most relevant to PD pathology is indicated in blue. (B) Medication details (additional information is presented in Table S1B). *Denotes a patient with record of cancer. Correlation of DNA methylation (average β-value) with age (C) or disease duration (D) plotted per individual PD subject studied. Blue circle identifies the patient with cancer record. Case ID corresponds with .
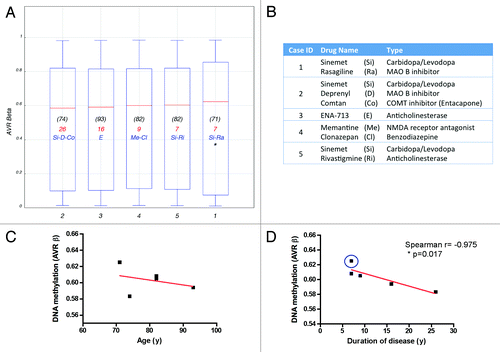
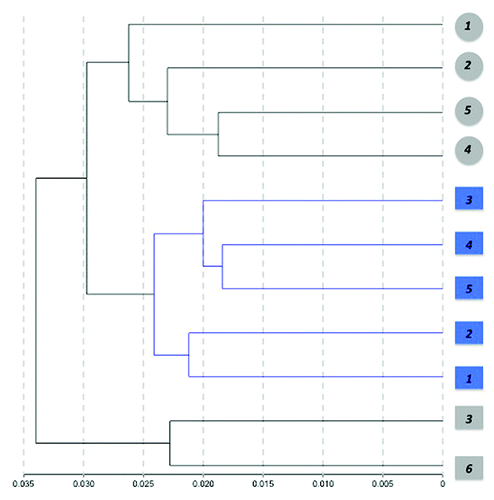
Figure 4. Hierarchical clustering of samples based on individual methylation profiles detected from blood. Dendogram showing the unsupervised hierarchical clustering of the 11 cohort subjects using absolute correlation. Control cases (gray) and PD patients (blue) were grouped based on Pearson correlations of whole genome methylation profiles using a 1 - |r| distance measure and nesting with average linkage method. An average of 485,386 CpG sites/group with detection p < 0.001 were featured in the analysis. Females are represented by circles and males by squares. Numbers represent the individual sample ID.