Figures & data
Table 1. Differentially methylated loci in ovarian and endometrial cancer
Figure 1. Methylation levels in tumors vs. normal controls for ovarian and endometrial cancer samples. For each probe, points associated with individual samples are drawn over the corresponding boxplots and violin-plots. The vertical axis shows the β value associated with each point, with values ranging from 0 (completely unmethylated) to 1 (completely methylated). Median values of methylation for each sample class are shown in .
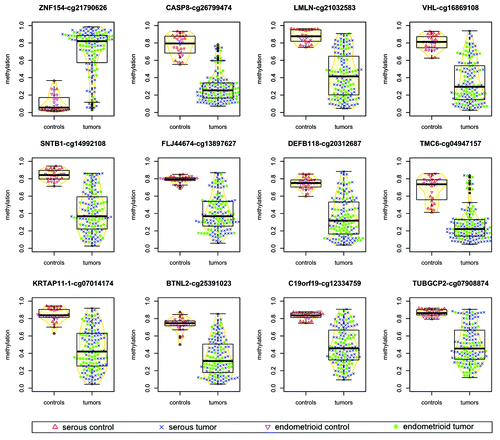
Figure 2. Methylation at the ZNF154 promoter is correlated with widespread methylation of CpG islands in endometrioid tumors. (A) Hierarchical clustering of all our ovarian and endometrial cancer samples using the 500 probes with the highest variance in methylation, selected as in Kolbe et al.Citation25 The heat map shows levels of methylation as β values associated with each pair of probe (columns) and sample (rows). Top horizontal color bar shows which of the probes are located within CpG islands. The vertical color bar on the left shows the level of methylation at cg21790626-ZNF154 for each individual sample. The vertical color bar on the right shows the sample class, including serous controls (SC), serous tumors (ST), endometrioid controls (EC), endometrioid tumors (ET) and papillary serous tumors of low malignant potential (LMP). (B) DNA methylation at cg21790626-ZNF154 vs. average DNA methylation at the set of 380 probes from the top panel that are located within CpG islands. Each point in the plot corresponds to an individual sample. (C) Distribution of P values for Spearman correlation coefficients between DNA methylation at cg21790626-ZNF154 and average methylation at sets of 380 probes that were randomly selected in CpG islands across the entire genome. Each of the two histograms shows results for one million random choices of probe sets.
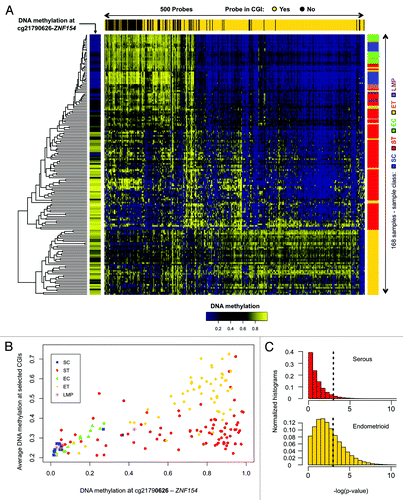
Table 2. Differentially methylated loci in ovarian and endometrial cancer and human cell lines
Figure 3. Seven of the 12 loci selected from our ovarian and endometrial cancer analysis show differential methylation in cancer vs. normal cell lines from ENCODE. The heat map shows β values that reflect the level of methylation associated with each probe (columns) in each cell line (rows). The color side bar on the left distinguishes cell lines that were derived from cancer (magenta), normal (green) or unspecified (black) tissues. Unsupervised hierarchical clustering based on pairwise correlations was used to order rows and columns. Median levels of methylation in cancer and normal cell lines are provided in .
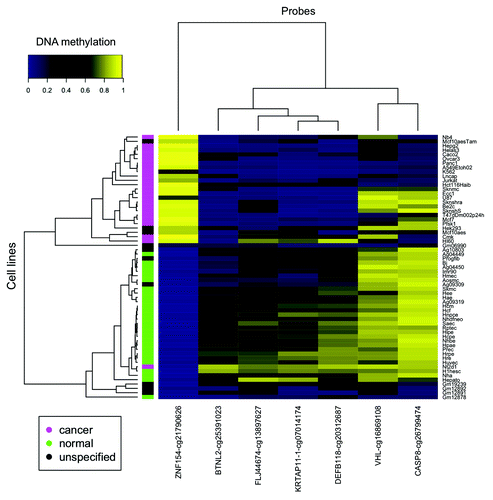
Table 3. TCGA data: types of cancer and number of methylation and expression data samples
Figure 4. Mean levels of methylation in tumors (red) vs. normal controls (blue) at the genomic locations shown in for 16 types of cancer from TCGA. Error bars show a 95% confidence interval centered at the mean. The number of samples used for each cancer type is provided in .
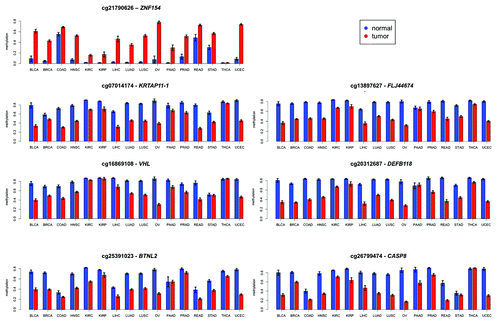
Table 4. Correlation between methylation and expression at differentially methylated loci
Figure 5. Expression of ZNF154 vs. methylation at probe cg21790626 in different cancer types from TCGA for tumors (red) and normal controls (blue). RNA-Seq data was used for the STAD type and RNA-Seq V2 was used for all the other types (see “Materials and Methods” for details). Combined methylation and expression data were not available for any normal controls of the COAD, OV, READ, and STAD types. Values of Spearman correlation, together with their associated levels of significance, are provided in .
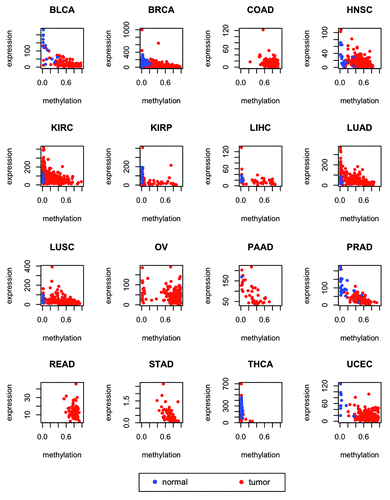
Figure 6. Methylation near the CpG island at chr19:58 220 189–-58 220 517, which is located at the promoter of gene ZNF154 and contains probe cg21790626 (highlighted in red). (A) Methylation levels in ENCODE cell lines depicted in the heat map show β values for each pair of probe (columns) and cell line (rows). Rows are ordered according to decreasing values of methylation at cg21790626 from top to bottom. The color side bar on the left shows cancer (magenta), normal (green), and unspecified (black) cell line annotations as provided by ENCODE. Probes located within the north shore (blue), island (orange), south shore (cyan), and south shelf (purple) are highlighted. The bar plot above the heat map shows the difference in median methylation in cancer cell lines minus median methylation in normal cell lines for each individual probe. (B) Difference in median methylation in tumors vs. controls at probes neighboring CpG island chr19:58 220 189–58 220 517 in different cancer types from TCGA. Ovarian cancer is excluded because no data from the Illumina 450K platform were available from TCGA at the time of our analysis. The color notation and the probe locations are the same as in the top panel. Positive values correspond to hypermethylation in tumors. Bars corresponding to probes that were marked as “not available” (NA) in level 3 data from TCGA (including the probe located within the north shore from panel A) are shown as empty horizontal gaps.
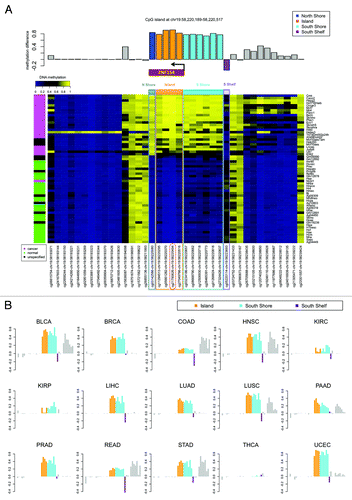
Figure 7. Methylation levels from our amplicon sequencing analysis, measured in K562 and GM12878 cell lines at individual CpGs on the forward strand of the genomic interval around the transcription start site of ZNF154 (chr19:58 220 404–58 220 705). (A) Percent methylation for each of the 20 CpG dinucleotides in the interval; CpGs are marked by their C-coordinates on chromosome 19. Methylation values from ENCODE data at probes from Illumina Infinium arrays within this region are shown for comparison purposes. (B) Distribution of reads as a function of methylated CpG fraction. K562 has most reads with 10 or more (≥50%) CpGs methylated, while the situation is reversed for GM12878.
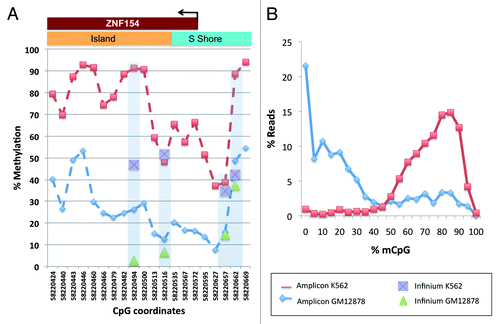
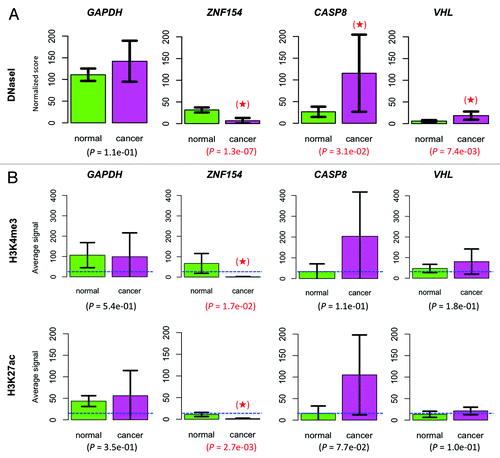
Figure 8. Chromatin accessibility at the ZNF154, CASP8, and VHL loci. (A) Comparison of DNaseI hypersensitivity signals in normal vs. cancer cell lines. Mean values were computed over 71 normal and 28 cancer cell lines. (B) Comparison of histone marks H3K4me3 and H3K27ac. In the ZNF154 case, mean values were computed over the entire CpG island. In the CASP8, VHL, and GAPDH cases, mean values were computed over a 52 bp region centered at the CpG dinucleotide interrogated by the corresponding Illumina probes. Results were averaged over 8 normal and 4 cancer cell lines from ENCODE. The mean value for each histone mark, averaged across all genes and cell lines, is provided as a background reference (dashed, blue horizontal line). In both panels, housekeeping gene GAPDH was included for control purposes, error bars show 95% confidence intervals for each estimated mean, P values are provided corresponding to a one-sided t-test comparison and significant results are highlighted with a red star (see “Materials and Methods” for details).