Figures & data
Figure 1 Genome-wide identification of genes associated with specific histone modifications in drug-resistant ovarian cancer cells. (A) Relative peak intensity for each mark. Target genes were identified using a peaksPicking program developed by Jin et al. (Genome Research 2006). Percentages of genes were calculated based on the total number of genes identified in the top 10% of genes from the Agilent array. A plot of each binding site based on the size of occupancy into a particular bin with an increment of 100 bp for each bin. (B) Histone H3 marks in Intragenic vs. promoter CpG islands (CpGIs). Number of individual peaks for each of the four H3 modifications examined (based on CpGIs featured on the array). The top 10% of the total hybridized microarray features (using peaksPicking software, see text for details) were used for the analysis. (C) Genes with H3 marks in proximity to transcriptional start sites (TSSs). Histone mark occupancies were predominately located in regions corresponding to −2 kb to 2 kb relative to the TSS.
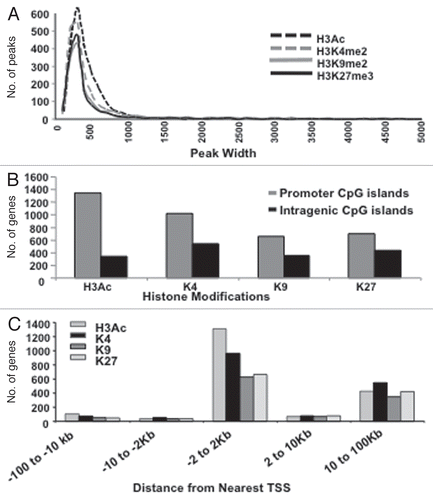
Figure 2 Histone marks and their various combinations, are redistributed or erased upon transition from 2D to 3D culture. PeaksPicking detection of monovalent, bivalent, trivalent and tetravalent H3 modifications in 2D and 3D cells. (A) Monovalent marks. All genes (the top 10%, as determined by the peaksPicking algorithm) were analyzed for the presence of individual histone marks. The proportion and type of such monovalently marked genes in 2D cells were compared to those in 3D cells. (B) Multivalent marks. All genes found by peaksPicking were analyzed for the presence of multivalent histone marks. The proportion and type of such multivalent genes in 2D cells were compared to those in 3D cells. (C) Distinctiveness and overlap of histone-marked genes between the 2D and 3D cellular states. Of the 2,734 genes detected by peaksPicking (top 10%) from 2D cells and 2,424 genes detected in 3D cells, 1149 were found to possess any of the four histone marks between the 2D and 3D.
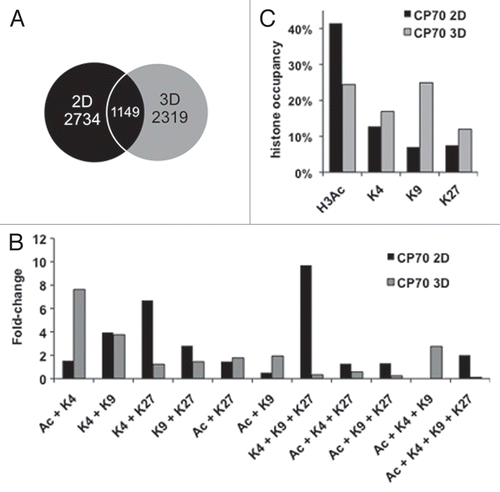
Figure 3 Multivalently histone-marked promoters, abundant in 2D cells, resolve largely to repressive monovalency under 3D culture conditions. (A) Both active and repressive monovalent promoters in 2D cells become mostly monovalent and repressive in 3D cells. All genes containing a monovalent mark in 2D cells were analyzed for the presence of histone marks in 3D cells. Inner pie is modifications less than 5% abundance. (B) Multivalent marks in 2D cells become predominantly monovalent in 3D cells. 2D cells contained 31% multivalent marks, which decreased to 21% multivalent marks in 3D cells. The 2D gene possessing the Ac + K4 + K9 mark was not marked in 3D cells. Black lines delineate monovalent marks from multivalent marks. All possible histone mark combinations are represented.
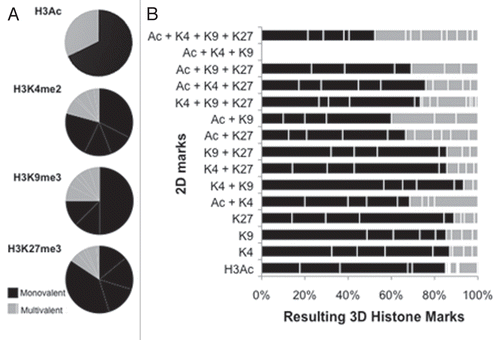
Figure 4 “Histone code”-based correlation of histone modification changes with changes in gene expression. (A) Box-plot analysis of gene expression and chromatin marks in 2D (left upper part) and 3D (right upper part) CP 70 cells. Gene expression arrays were performed on 2D and 3D CP70 cells and compared to those of nOSE cells, with changes in gene expression then correlated with specific chromatin marks associated with each gene. In accord with the histone code hypothesis, promoter-localized H3Ac and H3K4me2 correlated with gene upregulation, while H3K9me3 and H3K27me3 correlated with gene downregulation; genes having any two methylated H3 marks were mostly repressed. (B) DNA-binding, MMP and Wnt family genes are regulated by cellular architecture (2D vs. 3D). The most significant up-and-downregulated genes are shown.
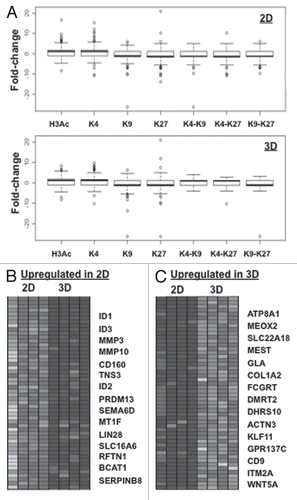
Figure 5 miRNAs are targeted during 2D to 3D transition. (A) miRNA microarray heat map. Top 15 upregulated and downregulated miRNA genes in CP 70-K27R cells, as compared to the parental CP 70 cells. A custom microarrayCitation84 was used to determine miRNA expression, using two replicates for each cell line. Clustering of miRNA expression data was performed using CLUSTER,Citation85 with filtering to remove inconsistencies between replicates. For clustering, we first log-transformed the data and median-centered the array and genes, followed by average linkage clustering. The expanded images flanking the heat map highlight the most significantly changed genes (p > 0.02, top 98%). No significant difference between replicated samples or between 2D and 3D for each cell line (p > 0.95).
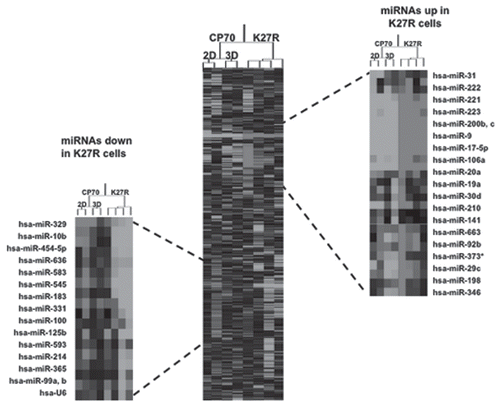
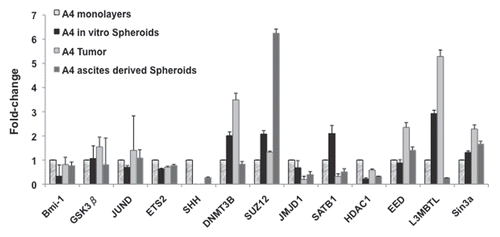
Figure 6 Flexibility of gene expression in xenografts. RT-qPCR results indicating flexibility of gene expression of monolayer CP 70 cultured cells vs. a primary sub-cutaneous tumor (A4 s.c) vs. spheroids in peritoneal metastases (A4 Sph-vivo). Q-PCR standard curves were produced in triplicate and Ct values used to determine relative expression analysis. The measured Ct value was further used to estimate the transcripts levels. Beta-actin was used as a reference housekeeping gene, with expression levels normalized to those of the same genes as expressed in normal ovarian surface epithelial (nOSE) cells.