Figures & data
Figure 1. Chemical structures of digoxin (A) and its cardioinactive metabolite, dihydrodigoxin (B). The double bond in the lactone ring (highlighted) becomes saturated, which reduces the affinity for its target, a Na+/K+ ATPase expressed in heart tissue.
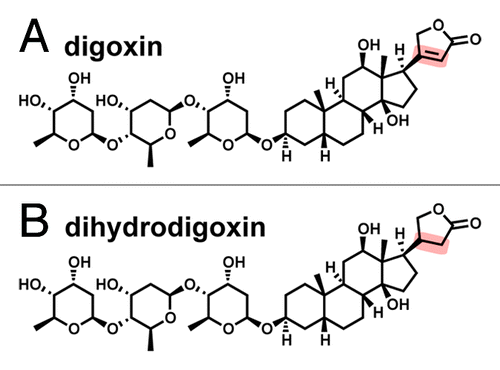
Figure 2. Predicted structures or Cgr1 and Cgr2, and their proposed interaction. (A) Genome mining revealed structural and sequence homology between the Cgr1 protein and several structurally characterized members the NapC/NirT family of cytochrome c reductases such as the NrfH enzyme from the Gram-negative bacterium Desulfovibrio vulgaris. This protein anchors as a dimer in the cytoplasmic membrane and shuttles quinone (Q)-derived electrons to associated periplasmic nitrite reductases (purple and gray). (B) Cgr2 exhibits homology to FAD-binding fumarate reductases and may serve as the terminal electron reductase partner to Cgr1 by forming an complex with Cgr1 and receiving electrons from Cgr1 at the active site FAD redox cofactor. (C) The structural and electronic similarities between the unsaturated dicarboxylic acid of fumarate and the α,β-unsaturated lactone of digoxin suggests that digoxin is able to occupy the active site of Cgr2 and undergo reduction by the Cgr1/Cgr2 complex.
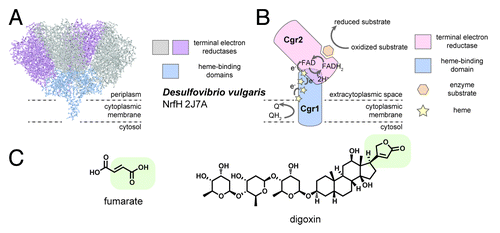
Figure 3. Approaches for studying the role of the microbiome in therapeutic drug metabolism. Initial evidence often comes from clinical data such as unexplained patient-to-patient variation in the response to therapeutics, and/or altered pharmacokinetics (PK) and pharmacodynamics (PD) in response to antibiotic treatment, dietary intake, IV vs. oral routes of drug administration, or varying oral formulations to delay absorption. Drug metabolites may then be identified directly from patient samples; from mouse or other animal models; or after ex vivo incubation with fecal samples. Mechanistic insight can be garnered by combining a number of complementary methods: functional metagenomics, microfluidics, and screening gut microbial communities for relevant enzymatic activities. Determining the signals that activate genes, and biochemical characterization of the relevant gene products, will enrich findings from these studies. From there, animal models will determine the translational potential, while human intervention trials could be utilized to work out co-therapy strategies.
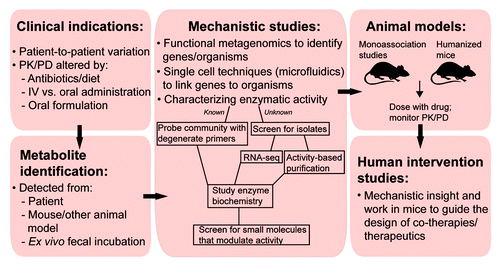
Figure 4. Integrating the gut microbiome into personalized or precision medicine. Rapid personalized clinical diagnostics may one day include ex vivo incubation of a patients’ microbiota with potential drug therapy cocktails, or “microbiota typing” using culture-dependent or -independent methods (e.g., sequencing, quantitative PCR). Patient populations might be stratified based on the results from these tests, and appropriate co-therapies may be administered. Potential therapeutic strategies include dietary supplements, prebiotics, probiotics, fecal microbiota transplantation, small molecule modulators of microbial gene expression/enzyme activity, or antibiotics.