Figures & data
Figure 1. Localization of phenylalanine residue 364 in the DNA-binding domain of STAT1 transcription factor. (A) Depicted is a ribbon diagram of the crystal structure of a truncated monomeric STAT1 molecule bound to DNA with the DNA-binding domain colored in yellow and the residue F364 in magenta.Citation25 (B) A closer view of the ribbon representation demonstrates the spatial orientation of the aromatic ring of F364 in relation to the residues Q340, G384 and Q408 (colored in blue), which are part of a pocket structure on the surface of the protein required for the interaction with the partner monomer in the antiparallel dimer conformation (not shown).
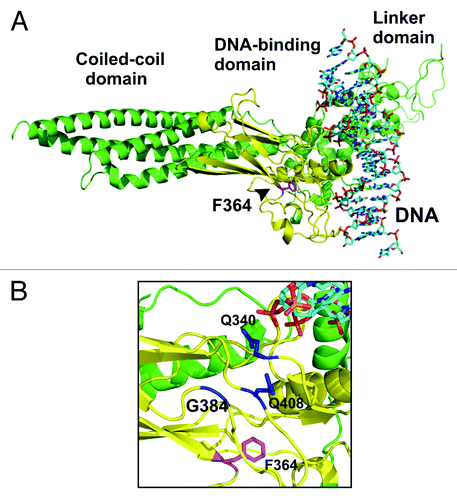
Figure 2. Substitution of alanine for phenylalanine at position 364 resulted in prolonged and elevated tyrosine phosphorylation of mutant STAT1 in cells stimulated with interferon-γ. (A–D) Equal cell numbers of HeLa cells expressing either wild-type (WT) or mutant STAT1-F364A, both tagged with green fluorescent protein (GFP), were exposed to IFNγ (2.5 ng/ml and 5 ng/ml, respectively) for 45 min before the kinase inhibitor staurosporine (500 nM) was added to the cells for the indicated times. Shown are the time courses of tyrosine phosphorylation as examined by western blotting using a STAT1-specific phospho-tyrosine antibody (αp-STAT1, top panel) and the same membranes after stripping and re-probing with the pan-STAT1 antibody C-24 (αSTAT1, bottom panel). Phosphorylation was monitored in whole cell lysates (A–C) as well as in cytosolic and nuclear extracts (D). The top arrowhead marks recombinant GFP-tagged STAT1 (S1-GFP) and the bottom arrowhead indicates endogenous STAT1 (S1). (B) Quantification of four independent western blotting experiments showing the ratio of phospho-STAT1 to total STAT1 normalized to STAT1-WT-expressing cells prior to adding staurosporine. Means and standard deviations for each time point are expressed. (E and F) Hyper-phosphorylation of STAT1-F364A did not result from decreased DNA binding. Cells expressing either F364A or DNAminus (V426D/T427D) were pretreated for 45 min with IFNγ and subsequently exposed to staurosporine for the indicated times. Cellular extracts were normalized to equal ratios of phosphorylated to total cellular STAT1 at baseline and loaded onto a SDS-gel. Shown are a representative western blot result (E) and a diagram depicting the decline in phospho-STAT1/STAT1 ratios for the two mutants [(F), means from two independent experiments]. The upper bands on each blot mark recombinant GFP-tagged STAT1, whereas the lower bands correspond to native STAT1.
![Figure 2. Substitution of alanine for phenylalanine at position 364 resulted in prolonged and elevated tyrosine phosphorylation of mutant STAT1 in cells stimulated with interferon-γ. (A–D) Equal cell numbers of HeLa cells expressing either wild-type (WT) or mutant STAT1-F364A, both tagged with green fluorescent protein (GFP), were exposed to IFNγ (2.5 ng/ml and 5 ng/ml, respectively) for 45 min before the kinase inhibitor staurosporine (500 nM) was added to the cells for the indicated times. Shown are the time courses of tyrosine phosphorylation as examined by western blotting using a STAT1-specific phospho-tyrosine antibody (αp-STAT1, top panel) and the same membranes after stripping and re-probing with the pan-STAT1 antibody C-24 (αSTAT1, bottom panel). Phosphorylation was monitored in whole cell lysates (A–C) as well as in cytosolic and nuclear extracts (D). The top arrowhead marks recombinant GFP-tagged STAT1 (S1-GFP) and the bottom arrowhead indicates endogenous STAT1 (S1). (B) Quantification of four independent western blotting experiments showing the ratio of phospho-STAT1 to total STAT1 normalized to STAT1-WT-expressing cells prior to adding staurosporine. Means and standard deviations for each time point are expressed. (E and F) Hyper-phosphorylation of STAT1-F364A did not result from decreased DNA binding. Cells expressing either F364A or DNAminus (V426D/T427D) were pretreated for 45 min with IFNγ and subsequently exposed to staurosporine for the indicated times. Cellular extracts were normalized to equal ratios of phosphorylated to total cellular STAT1 at baseline and loaded onto a SDS-gel. Shown are a representative western blot result (E) and a diagram depicting the decline in phospho-STAT1/STAT1 ratios for the two mutants [(F), means from two independent experiments]. The upper bands on each blot mark recombinant GFP-tagged STAT1, whereas the lower bands correspond to native STAT1.](/cms/asset/6a49bd16-b723-4fd5-8252-b9d55328ed58/kjks_a_10923576_f0002.gif)
Figure 3. The STAT1 mutant F364A shows unaltered kinetics of interferon-γ-induced nuclear accumulation. (A and B) Treatment of IFNγ-prestimulated HeLa cells with staurosporine led to a collapse of nuclear accumulation of GFP-tagged STAT1-F364A and restored its pancellular resting distribution with similar kinetics to the wild-type protein. Cells were either left untreated (− IFNγ) or treated for 45 min with 5 ng/ml IFNγ (+ IFNγ) before staurosporine (500 nM) was added for additional 0 and 60 min, respectively. (A) Microscopic images show the intracellular localization of STAT1-GFP and the Hoechst-stained nuclei. (B) The corresponding histogram demonstrates the nuclear-to-cytosol fluorescence intensity ratios with bars representing means and standard deviations. (C and D) Treatment with staurosporine for the indicated times resulted in a normal break-down of nuclear accumulation of untagged STAT1, which was independent of the presence of the F364A mutation. Immunocytochemical stainings using a STAT1-specific primary and Cy3-labeled secondary antibody show the rapid collapse of nuclear accumulation in IFNγ-prestimulated reconstituted U3A cells resulting from the exposure to staurosporine. Fluorescence microscopical images (C) demonstrate nuclear and cytoplasmic STAT1 concentrations in representative cells and the histogram (D) shows a quantitative analysis including means and standard deviations. The experiments were performed at least three times with similar results.
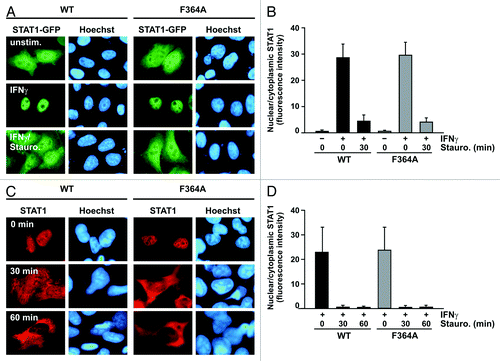
Figure 4. Decreased DNA-binding affinity of the STAT1 mutant F364A. (A) Gelshift experiment showing the reduced DNA-binding affinity of the mutant as compared with the wild-type protein. Before being loaded onto a native polyacrylamide gel, cell lysates from IFNγ-stimulated U3A cells (5 ng/ml) expressing either wild-type or mutant STAT1 were incubated with radioactively labeled DNA containing M67, GAS-nonGAS or 2 × nonGAS probes. The asterisk marks a non-specific band and the arrowhead at the right margin of the gel corresponds to STAT1. (B) Densitometric assessment of the DNA-binding activity of mutant and wild-type STAT1 normalized to equal amounts of phospho-protein as determined by immunoblotting (p = 0.032). (C and D) Comparison of the dissociation rate between STAT1-WT and F364A on a single STAT binding site (M67) as determined by electrophoretic mobility shift assay (EMSA). Whole cell extracts from IFNγ-prestimulated U3A cells were incubated with radioactively labeled DNA for 15 min and, subsequently, a 750-fold molar excess of unlabeled M67 was added for the durations indicated, before the samples were loaded onto a polyacrylamide gel. In the first lane, anti-STAT1 antibody C-24 was present in the EMSA reaction used for identification of STAT1-DNA complexes which are marked with an arrowhead. (E and F) Normal cooperative DNA binding due to tetramer stabilization of the F364A mutant. Extracts from an equal number of IFNγ-stimulated U3A cells expressing either wild-type or mutant STAT1 (5 μl in each lane) were incubated in vitro with [32P]-labeled DNA containing a tandem GAS site (2 × GAS). The reactions were either left unchallenged (-) or challenged for 30 min with a 750-fold excess of a single, unlabeled GAS site (+ Competition). (E) Autoradiography including, in lane 1, a supershift with 20 ng of STAT1 antibody C-24 added to the reaction. At the margin of the EMSA gel, the positions of tetrameric (T) and dimeric (D) STAT1 are marked with arrowheads. (F) Graphic representation showing the densitometrically measured signal intensities of dimeric-to-tetrameric STAT1 bound to 2 × GAS before (− Comp.) and after (+ Comp.) competition as determined from three independent experiments (p < 0.001). (G and H) Demonstration of the DNA-binding defect of the F364A mutant in HeLa cells expressing recombinant STAT1-NES-GFP, which coded for a transferable nuclear export signal (NES) situated between the cDNAs for full-length STAT1 and GFP. Cells expressing wild-type STAT1-NES-GFP or the respective F364A variant thereof were either left untreated or stimulated with 5 ng/ml of IFNγ in the presence or absence of 10 ng/ml of leptomycin B (LMB). Epifluorescence microscopic images of representative cells under the various stimulation conditions (G) and a quantitative analysis of the cytoplasmic/nuclear signal intensity ratios (H) are shown.
![Figure 4. Decreased DNA-binding affinity of the STAT1 mutant F364A. (A) Gelshift experiment showing the reduced DNA-binding affinity of the mutant as compared with the wild-type protein. Before being loaded onto a native polyacrylamide gel, cell lysates from IFNγ-stimulated U3A cells (5 ng/ml) expressing either wild-type or mutant STAT1 were incubated with radioactively labeled DNA containing M67, GAS-nonGAS or 2 × nonGAS probes. The asterisk marks a non-specific band and the arrowhead at the right margin of the gel corresponds to STAT1. (B) Densitometric assessment of the DNA-binding activity of mutant and wild-type STAT1 normalized to equal amounts of phospho-protein as determined by immunoblotting (p = 0.032). (C and D) Comparison of the dissociation rate between STAT1-WT and F364A on a single STAT binding site (M67) as determined by electrophoretic mobility shift assay (EMSA). Whole cell extracts from IFNγ-prestimulated U3A cells were incubated with radioactively labeled DNA for 15 min and, subsequently, a 750-fold molar excess of unlabeled M67 was added for the durations indicated, before the samples were loaded onto a polyacrylamide gel. In the first lane, anti-STAT1 antibody C-24 was present in the EMSA reaction used for identification of STAT1-DNA complexes which are marked with an arrowhead. (E and F) Normal cooperative DNA binding due to tetramer stabilization of the F364A mutant. Extracts from an equal number of IFNγ-stimulated U3A cells expressing either wild-type or mutant STAT1 (5 μl in each lane) were incubated in vitro with [32P]-labeled DNA containing a tandem GAS site (2 × GAS). The reactions were either left unchallenged (-) or challenged for 30 min with a 750-fold excess of a single, unlabeled GAS site (+ Competition). (E) Autoradiography including, in lane 1, a supershift with 20 ng of STAT1 antibody C-24 added to the reaction. At the margin of the EMSA gel, the positions of tetrameric (T) and dimeric (D) STAT1 are marked with arrowheads. (F) Graphic representation showing the densitometrically measured signal intensities of dimeric-to-tetrameric STAT1 bound to 2 × GAS before (− Comp.) and after (+ Comp.) competition as determined from three independent experiments (p < 0.001). (G and H) Demonstration of the DNA-binding defect of the F364A mutant in HeLa cells expressing recombinant STAT1-NES-GFP, which coded for a transferable nuclear export signal (NES) situated between the cDNAs for full-length STAT1 and GFP. Cells expressing wild-type STAT1-NES-GFP or the respective F364A variant thereof were either left untreated or stimulated with 5 ng/ml of IFNγ in the presence or absence of 10 ng/ml of leptomycin B (LMB). Epifluorescence microscopic images of representative cells under the various stimulation conditions (G) and a quantitative analysis of the cytoplasmic/nuclear signal intensity ratios (H) are shown.](/cms/asset/144a64f5-5110-435e-85f8-b1d6de8b7db5/kjks_a_10923576_f0004.gif)
Figure 5. Phosphorylated STAT1-F364A is partially resistant against inactivation by Tc45 phosphatase. (A–D) In vitro phosphorylation assays demonstrate unaltered tyrosine phosphorylation of STAT1-F364A. Whole cell extracts from reconstituted U3A cells expressing either STAT1-WT or -F364A (10 μl in each reaction) were incubated with 40 ng of recombinant JAK2 kinase (A and B) or 20 ng of EGF receptor [EGFR, (C and D)] and incorporation of phosphate in STAT1 was monitored with time by means of western blotting. Statistical analyses revealed no significant difference in the phosphorylation kinetics between wild-type and mutant STAT1 (p > 0.05). (E and F) STAT1-F364A is partially protected against the attack of the inactivating phosphatase, as revealed by an in vitro dephosphorylation assay. Extracts from IFNγ-prestimulated U3A cells (10 μl each) were incubated with 2 U of the STAT1-specific Tc45 phosphatase and tyrosine dephosphorylation was followed for 30 min. Shown are a representative western blot result (E) and a quantitative depiction (F) of the specific tyrosine phosphorylation (phosphotyrosine signal divided by total STAT1 signal) with bars expressing means and standard deviations. Significant differences between wild-type and mutant STAT1 from five independent experiments are indicated with asterisks (p = 0.034 and p = 0.017, respectively). (G) An electrophoretic mobility shift assay demonstrates the exchange of monomers between STAT1 dimers. Shown is a representative gelshift result using cellular extracts from U3A cells expressing either GFP-tagged or untagged STAT1 bound to M67 DNA. The identity of the bands corresponding to STAT1 (marked with arrowheads) was confirmed by the absence of or reduction in DNA-binding activity in IFNγ-stimulated Y701F- (lane 1) and unstimulated WT-expressing cells (lane 2) as well as from supershift reactions using either a STAT3- (lanes 3 and 5) or STAT1-recognizing antibody (lanes 4 and 6). In lanes 7–10, similar amounts of GFP-tagged and untagged homodimers were either immediately mixed and incubated together for 45 min (lanes 8 and 10) or incubated separately for 45 min before being loaded together onto the gel (lanes 7 and 9). The asterisk at the right margin marks a non-specific band.
![Figure 5. Phosphorylated STAT1-F364A is partially resistant against inactivation by Tc45 phosphatase. (A–D) In vitro phosphorylation assays demonstrate unaltered tyrosine phosphorylation of STAT1-F364A. Whole cell extracts from reconstituted U3A cells expressing either STAT1-WT or -F364A (10 μl in each reaction) were incubated with 40 ng of recombinant JAK2 kinase (A and B) or 20 ng of EGF receptor [EGFR, (C and D)] and incorporation of phosphate in STAT1 was monitored with time by means of western blotting. Statistical analyses revealed no significant difference in the phosphorylation kinetics between wild-type and mutant STAT1 (p > 0.05). (E and F) STAT1-F364A is partially protected against the attack of the inactivating phosphatase, as revealed by an in vitro dephosphorylation assay. Extracts from IFNγ-prestimulated U3A cells (10 μl each) were incubated with 2 U of the STAT1-specific Tc45 phosphatase and tyrosine dephosphorylation was followed for 30 min. Shown are a representative western blot result (E) and a quantitative depiction (F) of the specific tyrosine phosphorylation (phosphotyrosine signal divided by total STAT1 signal) with bars expressing means and standard deviations. Significant differences between wild-type and mutant STAT1 from five independent experiments are indicated with asterisks (p = 0.034 and p = 0.017, respectively). (G) An electrophoretic mobility shift assay demonstrates the exchange of monomers between STAT1 dimers. Shown is a representative gelshift result using cellular extracts from U3A cells expressing either GFP-tagged or untagged STAT1 bound to M67 DNA. The identity of the bands corresponding to STAT1 (marked with arrowheads) was confirmed by the absence of or reduction in DNA-binding activity in IFNγ-stimulated Y701F- (lane 1) and unstimulated WT-expressing cells (lane 2) as well as from supershift reactions using either a STAT3- (lanes 3 and 5) or STAT1-recognizing antibody (lanes 4 and 6). In lanes 7–10, similar amounts of GFP-tagged and untagged homodimers were either immediately mixed and incubated together for 45 min (lanes 8 and 10) or incubated separately for 45 min before being loaded together onto the gel (lanes 7 and 9). The asterisk at the right margin marks a non-specific band.](/cms/asset/cf95beab-4072-4d7d-bb61-cc74cee1792c/kjks_a_10923576_f0005.gif)
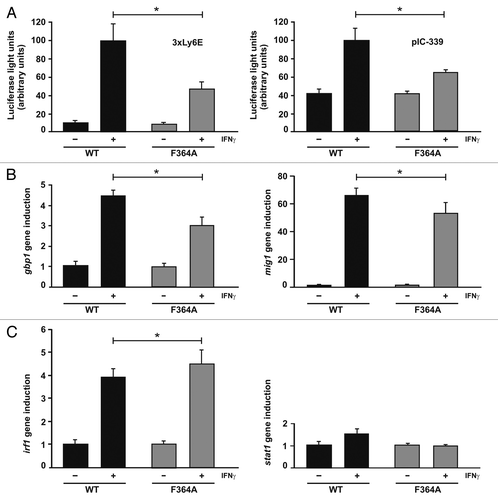
Figure 6. Transcriptional responses of the STAT1 DNA-binding mutant F364A. (A) Decreased reporter gene activation in U3A cells expressing STAT1-F364A as compared with the wild-type protein. U3A cells were transiently transfected with expression plasmids coding for either of the two STAT1 variants, the luciferase reporter constructs indicated and a constitutively expressed β-galactosidase gene used for normalization. The reporter constructs contained either a triple GAS site from the Ly6E promoter (3 × Ly6E) or a 339 base pair fragment from the native ICAM-1 promoter (pIC-339). On the next day, cells were either left untreated (− IFNγ) or stimulated for 6 h with 5 ng/ml of IFNγ (+ IFNγ) before, in whole cell extracts, luciferase luminescence and β-galactosidase activity were measured in six independent experiments. (B) Activation of three endogenous STAT1 target genes in U3A cells expressing either mutant or wild-type STAT1, as determined by real-time RT-PCR. Expression levels of the irf1, mig1, gbp1 and for control stat1 gene before and after 6 h stimulation with 5 ng/ml of IFNγ are shown. Gene induction was normalized to the expression of the house-keeping gene gapdh. The data are presented as means and standard deviations from at least three independent experiments. Statistical significance between the groups of IFNγ-stimulated cells expressing the indicated STAT1 variants is marked by asterisks.