Figures & data
Table 1. FcγR binding of analyzed antibodies
Figure 1. Glyco- or protein-engineered Fc variants optimized for FcγRIII binding mediated enhanced ADCC by MNC, but did not trigger PMN-mediated cytotoxicity. (A, B) ADCC against EGFR-expressing A431 cells was investigated in 3h 51chromium release assays in the presence of increasing concentrations of (A) protein-engineered or (B) glyco-engineered antibodies. ADCC by MNC was enhanced by protein- or glyco-engineered variants compared with wild type antibody (left panel). PMN triggered significant ADCC with wild type antibody, but were completely ineffective with both types of variants (right panel). Data are presented as mean ± SEM of at least three independent experiments with different donors. * P ≤ 0.05 for wild type vs. engineered antibodies. (C) FcγRIII-engineered antibody mediated tumor cell adhesion by PMN. A431 cell were co-incubated with GM-CSF stimulated PMN from healthy donors at an E:T ratio of 80:1 in the presence or absence of EGFR-targeted antibodies. After 3 h, microscopy was performed at 40x magnification (t = tumor target cells).
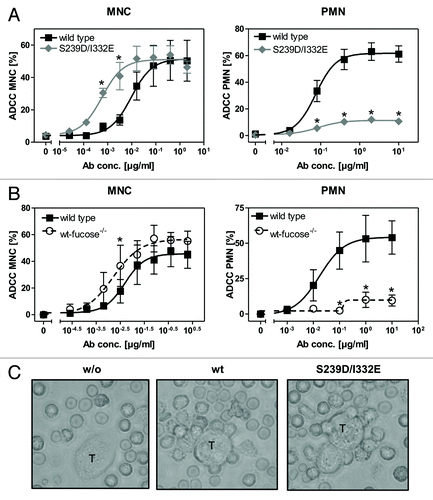
Figure 2. Impaired ADCC activity triggered by Fc-engineered antibody is restored using FcγRIII negative granulocytes. (A) Fc receptor expression was analyzed by indirect immunofluorescence. Unfractionated PMN from healthy donors (upper panel), patients with PNH (middle panel) or eosinophils (lower panel) were collected from freshly drawn peripheral blood. PMN from patients with PNH expressed low levels of the GPI-linked FcγRIII (CD16), while FcγRII (CD32) expression was similar to PMN from healthy donors. Unfractionated PMN from healthy donors could be divided into FcγRIII negative eosinophils and FcγRIII positive neutrophils (upper panel). FcγRIII negative eosinophils were isolated from the PMN fraction from healthy donors with eosinophilia and analyzed for FcR expression (lower panel). Eosinophils did express FcγRII (CD32) and FcαRI (CD89). (B) In ADCC experiments against A431 cells, PMN from healthy donors were effective with wild type but not with S239/I332E mutated anti-EGFR antibodies (left panel; all 10 µg/ml). PMN from PNH patients (middle panel) and eosinophils from healthy donors with eosinophilia (right panel) mediated similar ADCC with the S239D/I332E antibody variant and wild type antibody. For the three blood donor types, MNC-mediated killing was enhanced with Fc-engineered antibody (lower panel). In ADCC experiments against A431 cells, MNC-mediated killing was enhanced with Fc-engineered S239D/I332E antibody compared with wild type antibody (left panel). Data are presented as mean ± SEM from three independent experiments. * P ≤ 0.05 EGFR-targeted antibody vs. control antibody.
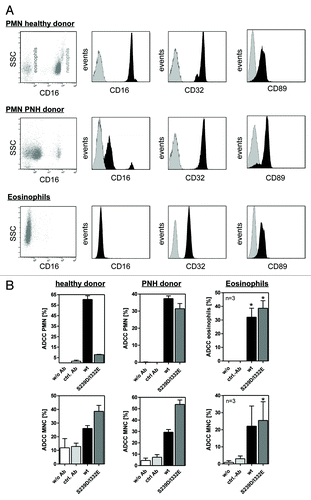
Figure 3. PMN-mediated ADCC is restored by blockade of FcγRIII. In ADCC experiments against A431 cells, PMN from healthy donors were effective with wild type, but not with S239D/I332E mutated anti-EGFR antibody (left panels), while MNC-mediated killing was enhanced with Fc-engineered compared with wild type antibodies (right panels). (A) FcγRIII blockade by anti-CD16 but not by the control antibody restored PMN-mediated ADCC activity (left panel) but impaired MNC-mediated ADCC activity (right panel) triggered by both antibody variants. (B) FcγRII blockade by anti-CD32 almost completely abolished PMN-mediated ADCC activity (left panel) for both antibody variants but did not affect MNC-mediated ADCC activity (right panel). Data are presented as mean ± SEM from three independent experiments with different blood donors. * P ≤ 0.05
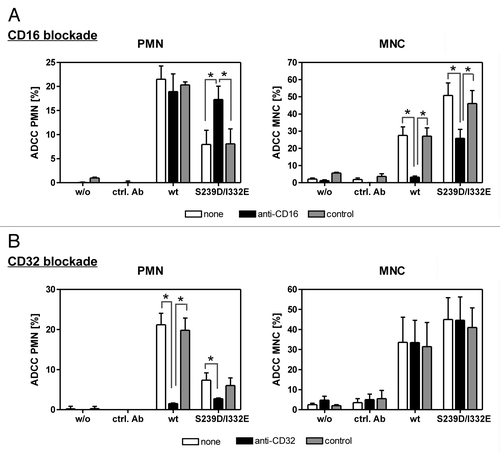
Figure 4. Determination of binding capacities of distinct antibody variants to FcγRIIIb. (A) Structural model of human IgG1 (pdb file from Clark, MR, Chem Immunol 1997; 65:88–110). The presented model illustrates the position of analyzed amino acid substitutions, which are located in the CH2 domain, as well as the fucose residues in IgG1. (B) Crystal structure of FcγRIIIb in complex with an Fc fragment of IgG1 (pdb file IT89). Pictures in A and B were generated using Discovery Studio 3.5 Client software (Accelrys). (C) SPR analyses were performed to determine binding capacities of distinct analyzed antibody variants to FcγRIIIb. One out of three independent experiments is presented per antibody variant. (D) Dose-response curves were prepared for analyzed antibody variants at indicated concentrations using CHO cell lines, either stably transfected with plasmids encoding for FcγRIIIb-NA1 or FcγRIIIb-NA2. Means ± SEM of at least three independent experiments are presented.
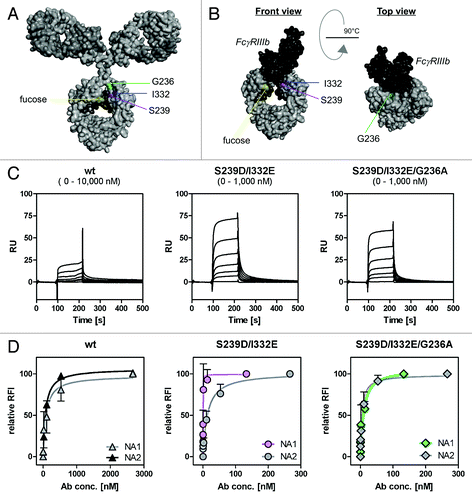
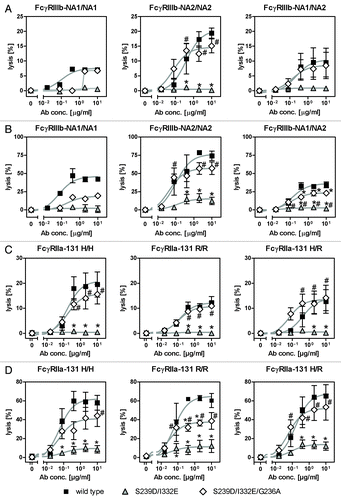
Figure 5. Impairment of PMN-mediated ADCC activity by an FcγRIII-optimized anti-EGFR antibody variant is restored by additional improvement of its FcγRIIa affinity. (A, B) PMN from healthy blood donors were left untreated (A) or were stimulated with GM-CSF (B) and subsequently used as effector cells in ADCC experiments utilizing indicated antibodies at increasing concentrations and A431 cells (left panels). Results from different blood donors at highest antibody concentrations were separately depicted (right panels). Data are presented as mean ± SEM from at least three independent experiments with different blood donors. * P ≤ 0.05 for wild type antibody vs. antibody variants; # P ≤ 0.05 for S239D/I332E/G236A vs. S239D/I332E. (C) Neutrophil-enriched whole blood samples from G-CSF primed donors were used as effector source in ADCC experiments against A431 cells. Data are presented as mean ± SEM from three independent experiments with different blood donors. * P ≤ 0.05 for wild type antibody vs. antibody variants; # P ≤ 0.05 for S239D/I332E/G236A vs. S239D/I332E.
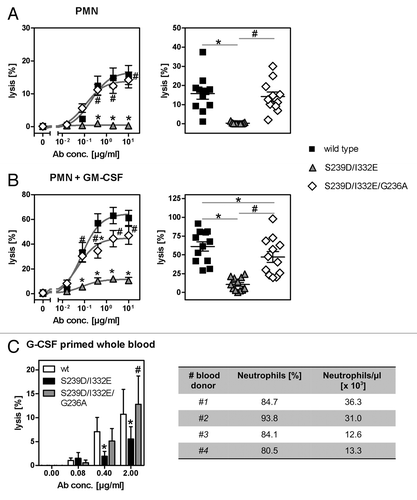
Figure 6. Enhancement of PMN-mediated ADCC by increased FcγRIIa binding is not limited to specific FcγRIIIb or FcγRIIa polymorphisms. (A-D) The effect of distinct FcγRIIIb (FcγRIIIb-NA1/NA2; A, B) and FcγRIIa (FcγRIIa-131H/R; C, D) polymorphisms on the extent of PMN-mediated ADCC against A431 cells by Fc-engineered anti-EGFR antibody variants was analyzed by 51chromium release experiments utilizing either untreated PMN (A, C) or GM-CSF stimulated PMN (B, C) from healthy blood donors. Data are presented as mean ± SEM from at least three independent experiments with different blood donors, except for FcγRIIIb-NA1/NA1 (n = 1). * P ≤ 0.05 for wild type antibody vs. antibody variants; # P ≤ 0.05 for S239D/I332E/G236A vs. S239D/I332E.