Figures & data
Figure 1. Genomic sequence comparison using the RAST Server (http://rast.nmpdr.org/). HMW 615 is compared with HMW 610, HMW 616, BF 9343, BF638R, and BF YCH46, respectively from perimeter of circle inward; HMW 615 sequence is inferred (not shown). The sequence of HMW 615 has been taken from the published supercontigs and rearranged according to the Mauve prediction. Hits on the comparison organisms are displayed graphically. Percent protein sequence identity is indicated (legend).
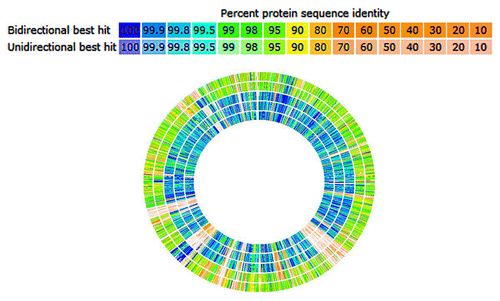
Figure 2. MAUVE-based alignment of BF 638R and BF-HMW615 and schematic indication of CTnHyb insertion into BF 638R. The MAUVE Contig Mover module was used to order (and orient) the BF-HMW615 supercontigs relative to the BF 638 reference genome. Colored block is a region of the genome that is homologous to a similarly colored part of another genome without extensive genomic rearrangement. Regions outside blocks lack detectable homology among the input genomes. Only a small part of the alignment is shown to visualize the insertion; nimJ and tetQ are indicated for orientation.

Figure 3. (A) Predicted events leading to integration of CTnHyb into the BF638R chromosome to result in BF638R/CTnHyb. The circular form of CTnHyb recombines with BF638R at the underlined sequence (GAAAGTGAA). (B) Alternate integration of CTnHyb into the BF638R chromosome. Predicted events leading to integration of CTnHyb into the BF638R chromosome to result in BF638R/CTnHyb found in HMW 876 and HMW 879 . The circular form of CTnHyb recombines with BF638R at the underlined sequence (TTTGA). (C) Predicted model of the deletions in CTnHybL leading to CTnHybS. The locus tag labels serve as approximate reference points. The bases are referred to as “bases” or “b”. The regions of homology that are predicted to recombine are color coded. The nimJ gene is among the genes deleted in the first deletion and metronidazole has a lower MIC for BF638R/CTnHybS than for BF638R/CTnHybL, as expected ().
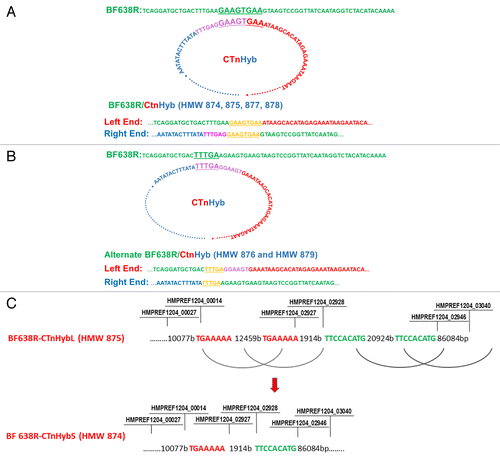
Figure 4. CTnHyb: Circularized form and predicted nested ICEs. Circular intermediates were detected in HMW 615, and in 4 isolates of BF638R/CTnHybL using CTnHyb/BF 638R Junction primers at either end of CTnHyb (Primers 2783 and 2784). The continuous sequence was generated using 4 GenBank files consisting of 1) the known Broad sequence within supercontig 1.1, an unannotated transpose downstream of the predicted HMPREF1204_0001, a short nucleotide segment and the sequence within supercontig 1.3 (we obtained the 2nd and 3rd sequence from manual PCR and sequencing). The GenBank files were concatenated using the SeqNinja program (DNA Star) and the generated sequence was annotated and visualized with the SeqBuilder program (DNA Star). The inner most circle is the GC% (green is higher than average BF GC % and purple is lower than average BF GC %). The predicted attachment site, the nimJ, mefA and tetQ genes and the predicted nested ICEs are indicated on the figure. 1) CTnHyb “Popout 1”: (missing in CTnHybS) 12466 bases long, extending from HMPREF1204_00014 to HMPREF1204_00001 of contig 1.1 and the unannotated transposase and HMPREF1204_02924 to HMPREF1204_02926 of contig 1.3); 2) NimJ cassette, 9955 bases, present in 3 copies in BF HMW 615; 3) Cassette (minus nimJ) is found in other BF strains; 3) nimJ gene; 4) CTnHyb “Popout 2 (missing in CTnHybS): 20,933 bases long; extending from HMPREF1204_02928 to HMPREF1204_02946; 5) The “foreign segment” extends from HMPREF1204_2965 to HMPREF1204_2980; 6) mefA (HMPREF 1204_02965, 1232 bp) is a 95% match to mefA genes on ICEs from Staphylococcus, Streptococcus, and EnterococcusCitation10, and a close mefA homolog was also found in CTnGermCitation11; 7) Eubacterium ventriosum cassette (7178 bp not counting the kanamycin cassette within); 8) kanamycin cassette (1517 bp); HMPREF1204_2969-HMPREF1204_2971 is 100% match to nucleotide regions on ICEs from Streptococcus. However, the transposase (HMPREF 1204_02971) in the “kanamycin cassette” is not homologous to the Staphylococcus or Enterococcus transposases, but is hom0logous to a transposase from Blautia hansenii—a novel genus of Gram-positive, anaerobic, non-sporulating coccobacillus-shaped bacteria that includes several former coccobacillary shaped species of Clostridia and Ruminococcus.Citation12 HMPREF 1204_02971 is truncated at a 10 bp palindrome (ACTTCCGCCG, bp 195–204 and 218–227 within HMPREF 1204_2970) which is characteristic of transposase insertions. Bacterial repetitive extragenic palindromic sequences are known DNA targets for Insertion Sequence elements.Citation13 This palindrome, however, is contained within the coding region of HMPREF1204_02971.
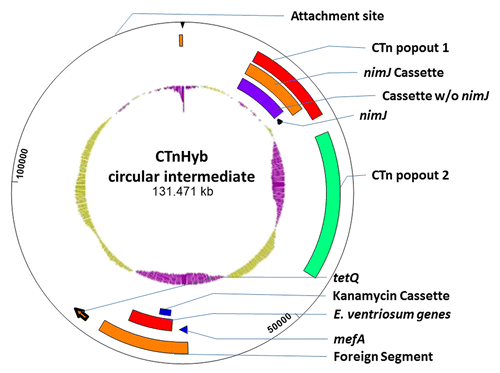
Figure 5. Distribution of genes in CTnHyb according to COG (Cluster of Orthologous Gene class). COG ID numbers and classification were taken from the Integrated Microbial Genomes and Metagenomes (IMG) at the Department of Energy Joint Genome Institute. (http://img.jgi.doe.gov/cgi-bin/w/main.cgi?section=FindGenes&page=geneSearch). Eighty-eight genes in CTnHyb do not belong to any COG. Similarly, RAST analysis of the entire BF-HMW 615 genome sequence indicates that 69% of the putative genes are not assigned to any subsystem (a subsystem is a method of categorization that can be thought of as roughly equivalent to a COG). Of the remaining 57 genes in CTnHyb(L), almost one third of the genes (n = 18) are in COG L (replication, recombination and repair) which includes such genes as DNA primases, excionases, integrases and transferases. Another 11 genes are annotated as “viral proteins” and are not assigned to a COG class, even though several are also functionally annotated as excisionase or integrase proteins.
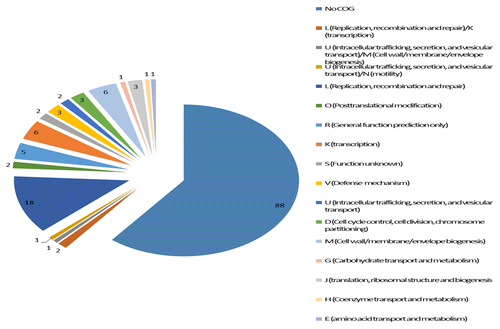
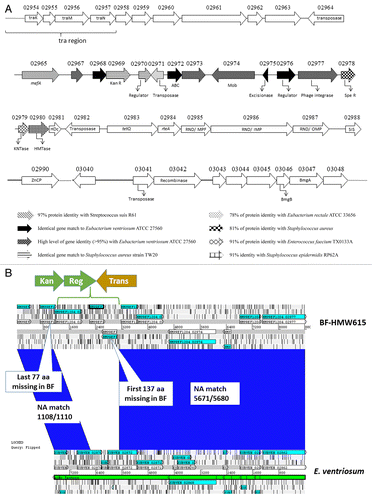
Figure 6. (A) Schematic representation of the “foreign” segment in CTnHyb. White arrows correspond to conserved genes in Bacteroides sp strains. See the figure for explanation of other arrows. APH(3′): aminoglycoside 3′-phosphotransferase; ABC: ABC transporter ; Mob: mobilization protein; Spe R: spectinomycin adenylyltransferase; KNTase: nucleotidyltransferase; HMTase: methlytransferase; HDc: Metal dependent phosphohydrolases with conserved “HD” motif; SIS: Sugar Isomerase; ZnCP: Zinc peptidase; BmgA and BmgB: mobilization proteins. (B) ACT alignment of area of homology between Eubacterium ventriosum and CTnHyb (BF-HMW 615). A segment (7671 bp) of the E. ventriosum genome is nearly completely conserved except for a three gene insert that replaces part of EUBVEN_02872, EUBVEN_02873 and part of EUBVEN_02874. In the conserved portions, only 11 nucleotides differed.