Figures & data
Figure 1. Ribbon diagram of the cellular prion protein (PrPC) showing the N-terminal octarepeat domain (gold), the folded C-terminal domain (blue), and the central hydrophobic region (red). Note that residues 23–55 are unstructured and omitted for clarity. The 4 His residues (magenta) in the octarepeat domain coordinate zinc and copper ions, as shown. The additional His residues at 95 and 110 are also capable of coordinating Cu2+. Previously proposed sites for α-cleavage β-cleavage are indicated at the beginning of the hydrophobic segment and the end of the octarepeat domain, respectively.
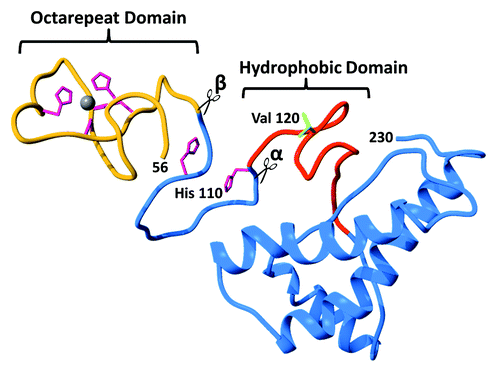
Figure 2. Global PrPC fold promoted by Zn2+, as demonstrated by Spevacek et al. using NMR and double label electron paramagnetic resonance.Citation25 The Zn2+ occupied octarepeat domain makes a tertiary contact with the surface formed by C-terminal helices 2 and 3. Note that helices 2 and 3 present a cluster of negatively charged residues that facilitate the tertiary contact, and that familial mutations weaken the fold.
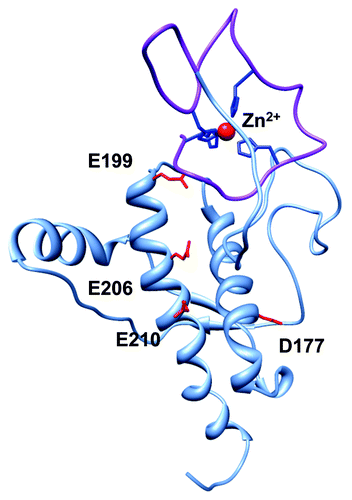
Figure 3. Summary of the findings of McDonald et al. on the α-cleavage and β-cleavage by ADAM family enzymes.Citation8 N-terminal (white) and C-terminal (green) domains are shown, with octarepeats (gold), as is the C-terminal GPI moiety that anchors the protein to the extracellular membrane leaflet. (A) ADAM8, ADAM10, and ADAM17 produce α-cleavage at distinct sites, noted as α1, α2, and α3. ADAM10 also acts as a sheddase by cleaving near the C-terminus to release the protein from the membrane. Also shown are the sites in the octarepeat domain where ADAM8 produces β-cleavage. (B) Relative cleavage activities are indicated by arrow thickness. ADAM8 alone (no metal ions) produces extensive α1- and β-cleavage, with α2-cleavage as a minor product. (C) In the presence of Cu2+, α1- and β-cleavage are suppressed, while α2-cleavage is enhanced. (D) Zn2+ also inhibits β-cleavage but, in contrast to Cu2+, promotes α1- over α2-cleavage. In contrast to the previous paradigm, these findings show that metal ions suppress β-cleavage and template a structure that promotes α-cleavage.
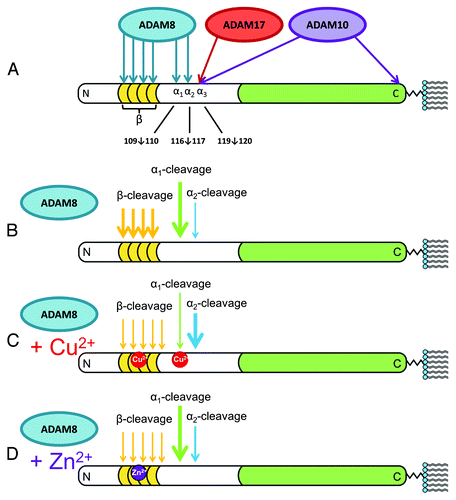
Figure 4. Summary of PrPC deletion mutants and resulting phenotypes (sequence details summarized in Aguzzi et al. ref. Citation6). The N-terminal domain after removal of the signal peptide starts at residue 23, and is shown in blue, while the structured C-terminal domain, starting at approximately residue 125 is shown in green. Deleted segments, as noted by the numbering (left), are gray. Locations for α1, α2, and α3-cleavage are shown. The green checks, right, indicate tolerated deletion mutants, whereas the red X’s are the lethal mutants. The number of Xs qualitatively represents the lethal potency of each mutant, as indicated by the relative amount of wildtype PrPC required for rescue. This alignment shows that deletion of the full α-cleavage segment, 109–120, but with retention of N-terminal residues starting at position 23 confers toxicity.
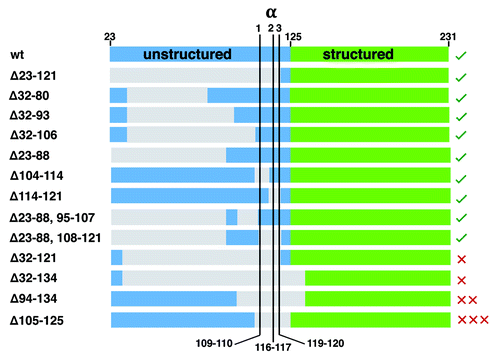
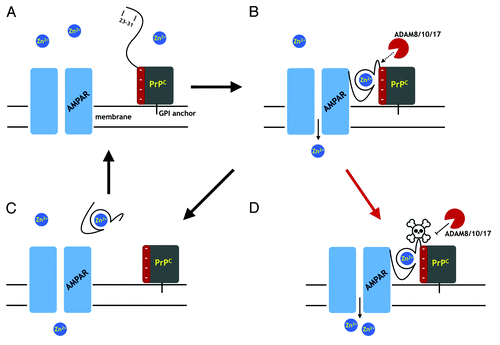
Figure 5. A proposal for PrPC regulation through α-cleavage. (A) The AMPA receptor (AMPAR), a cation channel, is one of several membrane proteins thought to be modulated by PrPC binding. The folded PrPC C-terminal domain (square) is GPI linked to the extracellular membrane. The N-terminal domain is unstructured in the absence of Zn2+. (B) Zn2+ binds to the octarepeats, structuring the N-terminal domain, which then associates with a negatively charged patch on the C-terminal domain. Residues 23 – 31 bind to the AMPAR facilitating Zn2+ transport. The PrPC fold promoted by Zn2+ exposes the α-cleavage site, which becomes susceptible to the ADAM proteases (8, 10, and 17). (C) α-Cleavage releases N1, thus downregulating PrPC facilitated Zn2+ transport. C-Terminal PrPC (the C1 fragment) is degraded and replaced by full-length PrPC. (D) Occlusion of α-cleavage (skull and crossbones) by the peptide PrP(106–126) or Aβo, for example, or by misfolding to PrPSc, leaves PrP in a constitutively active state, thus driving dyshomeostasis.