
Abstract
Background
Fabry’s disease is an X-linked lipid storage disorder due to deficient lysosomal alpha galactosidase A.
Case Presentation
Kidney biopsy was done on a 19 year old male patient with complaint of acroparesthesia, maculopapular skin lesions and cornea verticillata. Kidney biopsy tissue was processed and examined by electron microscopy. Changes were inclusion bodies in the cytoplasm of the renal cells. These inclusions were osmophilic with concentric lamellation of clear and dark layers, showing onion skin appearance. The podocytes were mostly affected and some of the foot processes were fused. Cross-sections of collagen fibers were also evident, indicating fibrosis.
Conclusion
The ultra-structure of the kidney clearly showed the intra-cytoplasmic glycosphingolipid accumulation in renal cells, responsible for progressive decline in renal function which could lead to kidney failure. The final diagnosis of Fabry’s disease was confirmed. In the present case-study, electron microscopy proved to be a valuable diagnostic aid.
1 Introduction
Fabry’s disease is an X-linked lipid storage disorder due to deficient lysosomal alpha galactosidase A. In this disease dermatological and soft tissue symptoms, angiokeratomas are common skin lesions which appear in adulthood and are considered as a significant diagnostic value.Citation1 Cornea verticillata was noted in 94.1% hemizygotes and 71.9% heterozygotes patients and considered as an ophthalmological indicator of this disease.Citation2 Globotriaosylsphingosine was identified as a bioactive molecule accumulating in Fabry’s disease, which could modulate the release of secondary mediators of injury in podocytes.Citation3 High doses of agalsidase showed to have effects on the clearance of inclusion bodies in epithelial cells of distal tubules as well as podocytes.Citation4 Incidence of the disease has been estimated to range from one per 40,000 to one per 117,000 male live births. The gene for this disorder is on the X-chromosome and the mother needs to be a carrier to produce an affected child.Citation5,Citation6 Mean survival is 40–50 years for males and 70 years for female carriers, death occurs from cardiac, renal, or cerebrovascular complications.Citation7 Kidney is affected in all hemizygous male. Renal pathological and functional impairment is more evident in hemizygous males than heterozygous females.Citation8 Studies revealed greater podocyte vacuolization in male patients than in female ones.Citation9 Progressive intracellular deposition of glycosphingolipid which ultimately leads to end-stage renal failure.Citation10–Citation12 By light microscopy, distinctive “Foamy” cytoplasmic alterations were observed in renal glomerular, tubular, vascular, and interstitial cells.Citation13–Citation15 Large amounts of lipid material are seen in podocytes followed by parietal epithelial, mesengial and glomerular endothelial cells.Citation16 Immunohistochemical localization of glycosphingolipid was also documented.Citation17 Other methods failed to reveal small amounts of stored glycolipid.Citation18 Kidney biopsy is of importance in evaluation of Fabry nephropathy.Citation9 Maltese cross particles, anti-CD77 antibody binding within vacuolated urinary epithelial cells were detected in Fabry disease.Citation19 Large numbers of electron dense deposits in the renal tubules was reported,Citation20 these deposits showed characteristic “onion skin” or “zebra appearance” in all kinds of renal cells.Citation15 Electron microscopic examination of kidney biopsy specimen is important for investigation of storage diseases.Citation21
2 Case report
Kidney biopsy was done on a 19 year old male patient with complaint of acroparesthesia, maculopapular skin lesions and corneal verticillata. Various investigations preformed on the patient are presented in , based on overall results, kidney biopsy was performed, tissues fixed and processed routinely and ultrathin sections of 60–70 nm were cut, stained with uranyl acetate and lead citrate and examined under TEM (Philips CM 100). The ultrastructural changes were mainly intracytoplasmic inclusion bodies in the renal cells, they were dense osmophilic with concentric lamellation of clear and dark layers, showing onion skin, zebra appearance and sometimes fingerprint like myelin figures (A). These electron dense deposits were more in the podocytes (B and A), they were also seen in the mesangial cells (B), and were found to be less in the tubular epithelial cells. Podocytes were most affected in these structures. In some places foot processes were found to be fused. Cross-sections of collagen fibers were also evident in different parts, indicating fibrosis.
Fig. 1 A – Electron micrograph of intracytoplasmic osmophilic inclusion body (Arrow), showing fingerprint-like myelin figures (Original magnification ×13,500). B – Dense osmophilic inclusion bodies (I) packed in the cytoplasm of a podocyte, capillary lumen (C) is seen (original magnification ×1850).
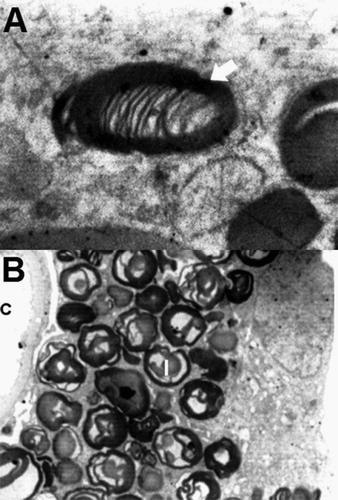
Fig. 2 A – Electron micrograph of a podocyte (P) with osmophilic inclusion bodies (I) in its cytoplasm which are typical onion skin or zebra appearance, basement membrane (BM) and capillary lumen (C) are also seen (Original magnification ×7400). B – Mesangial cell (M) with dense osmophilic inclusion bodies (I) in its cytoplasm (Original magnification ×3400).
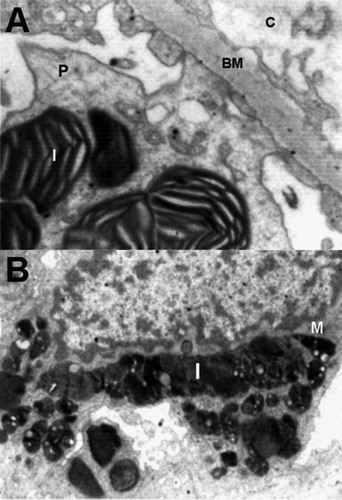
Table 1 Summary of investigations.
3 Discussion
Intra-cytoplasmic accumulation of glycosphingolipid in the renal cells was clearly demonstrated, they were dense osmophilic concentric lamellar structures that would lead to kidney failure. Myelinosomes in great numbers were found in certain inborn errors of metabolism, these bodies have been given various names such as lipid cytosomes, multimembranous bodies, lamellar bodies, membranous cytoplasmic and zebra bodies. Myelinosomes containing stacked membranes and membranous whorls have been reported in kidney epithelial cells. Both the types of zebra bodies and whorl types were seen, which they were of zebra body types predominantly. In this case we observed most of these bodies in the podocytes and mesengial cells respectively and very minute in the tubular epithelium in contrast to the other reports in which large amounts of electron dense material was seen in the renal tubules.Citation20 This could be the reason in which we could not detect vacuolated epithelial cast in the urine analysis. Dysregulated autophagy in alpha-galactosidase A deficient mTOR kinase activity, which lead to deregulated autophagy pathways, opened a new horizon on pathogenesis of glomerular injury in Fabry disease.Citation21,Citation22 Electron-microscopy is a far superior and easier way to diagnose these bodies than any other technique, however, light microscopy in which cytoplasmic changes in renal cells could be due to other lipid material could not be differentiated from glycosphingolipid,Citation13–Citation15 on the other hand, when the amount of glycosphingolipid is minute, even histochemistry will not be of diagnostic value.Citation18 In the present case-study, electron microscopy proved to be a valuable diagnostic aid.
4 Conclusion
The ultra-structure of the kidney clearly showed the intra-cytoplasmic glycosphingolipid accumulation in renal cells, responsible for progressive decline in renal function with which the final diagnosis of Fabry’s disease was confirmed. In the present case-study, electron microscopy proved to be a valuable diagnostic aid.
Conflict of interest
Author states that there is no conflict of interest.
Acknowledgement
The authors wish to thank Mr. Khashayar Farshid for his extensive language editing.
Notes
Peer review under responsibility of Alexandria University Faculty of Medicine.
Available online 23 June 2017
References
- P.C.LunaP.BoggioM.LarraldeDermatologic aspects of Fabry diseaseJ Inborn Errors Metab Scr4201617
- T.T.NguyenT.GinK.NichollsA.CrawfordOphthalmological manifestations of Fabry disease: a survey of patients at Royal Melborne Fabry disease CentreClin Exp Ophthalmol332005164168
- M.D.Sanchez-NinoA.B.SanzS.CarrascoGlobotriaosylsphingosine actions on human glomerular podocytes: implications for Fabry nephropathyNephrol Dial Transplant26201117971802
- G.TondelL.BostadK.K.LarsenAgalsidase benefits renal histology in young patients with Fabry diseaseJ Am Soc Nephrol242013137148
- P.KesN.Basic-JukicKidneys in Anderson-Fabry diseaseActa Clin Croat4420055963
- H.C.ChenJ.H.TsaiY.H.LaiJ.Y.GuhRenal changes in heterozygous Fabry’s disease – a family studyAm J Kidney Dis151990180183
- R.J.DesnickR.BradyJ.BarrangerA.J.CollinsFabry disease, an under-recognised multisystemic disorder:expert recommendation for diagnosis, management, and enzyme replacement therapyAnn Intern Med1382003338346
- M.F.LyonGene action in the X-chromosome of the mouse(Mus musculus L.)Nature1901961372373
- A.B.FogoL.BostadE.SvarstadScoring system for renal pathology in Fabry disease: report of International Study Group of Fabry nephropathy (ISGFN)Nephrol Dial Transplant26201021682177
- A.SessaM.MeroniG.BattiniEvolution of renal pathology in Fabry diseaseActa Paediatr Suppl92200368
- M.MiuraY.TominoW.InoueA case of Fabry’s diseaseTokai J Exp Clin Med819832329
- F.A.Reyes MarinB.Gomez NavarroY.TamayoNephropathy in a case of Fabry’s diseaseRev Invest Clin431991373376
- P.M.BurkholderS.J.UpdikeR.A.WareClinicopathologic, enzymatic, and genetic features in case of Fabry’s diseaseArch Pathol Lab Med10419801725
- M.A.IdoateF.J.Pardo-MindanAlamillo C.GonzalezFabry’s disease without angiokeratomas showing unusual eccrine gland vacuolationJ Pathol16719926568
- M.MatsubaraY.TagumaT.SaitoK.YoshinagaT.FuruyamaH.SakaguchiThe possible role of degenerative lesions in the interstitial vessels in renal dysfunctionNippon Jinzo Gakkai Shi321990237243
- J.AloryS.SabnisJ.B.KoppRenal pathology in Fabry diseaseJ Am Soc Nephrol13suppl 22002134138
- S.ChatterjeeP.GuptaR.E.PyeritzP.O.KwiterovichJr.Immunohistochemical localization of glycosphingolipid in urinary renal tubular cells in Fabry’s diseaseAm J Clin Pathol8219842428
- A.VoglinoM.ParadisiG.DompeA.Onetti MudaT.FaraggianaAngiokeratoma corporis diffusum(Fabry’s disease) with unusual features in a female patient. Light- and electron-microscopic investigationAm J Dermatopathol101988343348
- M.SelvarajahK.NichollsT.D.HewitsonG.J.BeckerTargeted Urine microscopy in Anderson Fabry disease: a cheap, sensitive and specific diagnostic techniqueNephrol Dial Transplant26201131953202
- T.SuzukiT.FujinoM.SugasawaA case of Fabry’s disease with chronic renal failureNippon Jinzo Gakkai Shi411999448453
- M.Wagrowska-DanilewiczM.DanilewiczZ.GozdzikThe ultrastructural changes in renal biopsy compatible with Fabry’s diseasePol J Pathol5019996163
- M.C.LiebauF.BraunK.HopkerC.WeitbrechtV.BartelsR.U.MullerDysregulatory autophagy contributes to podocyte damage in Fabry’s diseasePLoS ONE82013e63506 10.1371