
Abstract
Objectives: To identify the role of next-generation sequencing (NGS) in male infertility, as advances in NGS technologies have contributed to the identification of novel genes responsible for a wide variety of human conditions and recently has been applied to male infertility, allowing new genetic factors to be discovered.
Materials and methods: PubMed was searched for combinations of the following terms: ‘exome’, ‘genome’, ‘panel’, ‘sequencing’, ‘whole-exome sequencing’, ‘whole-genome sequencing’, ‘next-generation sequencing’, ‘azoospermia’, ‘oligospermia’, ‘asthenospermia’, ‘teratospermia’, ‘spermatogenesis’, and ‘male infertility’, to identify studies in which NGS technologies were used to discover variants causing male infertility.
Results: Altogether, 23 studies were found in which the primary mode of variant discovery was an NGS-based technology. These studies were mostly focused on patients with quantitative sperm abnormalities (non-obstructive azoospermia and oligospermia), followed by morphological and motility defects. Combined, these studies uncover variants in 28 genes causing male infertility discovered by NGS methods.
Conclusions: Male infertility is a condition that is genetically heterogeneous, and therefore remarkably amenable to study by NGS. Although some headway has been made, given the high incidence of this condition despite its detrimental effect on reproductive fitness, there is significant potential for further discoveries.
Overview of next-generation sequencing (NGS)1 technologies in the study of genetic disease
Genetic investigation of human populations has made remarkable advances in recent years, owing to the development and availability of NGS platforms. In contrast to the laborious process of single-gene mutation screening through exon-by-exon amplification and Sanger sequencing, NGS enables the interrogation of large panels of genes in a single experiment and at a reasonable cost [Citation1Citation[2]–Citation3].
NGS can be broadly classified into two categories: targeted panels or whole genome. Targeted methods (sometimes also referred to as ‘panel sequencing’) include investigation of a group of genes (referred to as a ‘gene panel’), usually selected on the basis of known disease association, or expanded to include genes within known disease pathways. Commercially produced custom-capture panels may be tailored to fit any number of genomic fragments of interest. The most comprehensive panel approach is therefore whole-exome sequencing, in which all coding regions are captured and sequenced. Typical whole-exome sequencing panels also capture flanking regulatory regions, enabling assessment of mutations affecting conserved but non-coding genic elements, e.g. splice junctions and 3′ and 5′ untranslated region (UTR) sequences [Citation4].
Beyond whole-exome sequencing, whole-genome sequencing is used to discover variants in the entire human genome. Alongside the advantage of covering non-coding and inter-genic regions, whole-genome sequencing does not require target enrichment prior to sequencing, and thus is possible with minimal sample preparation and results in sequenced fragments that appear evenly distributed across all chromosomes. This random distribution results in similar coverage across most of the genome, which means that variants can be reliably called at average genome depth as low as 20×. This is contrary to panel-based (e.g., whole-exome sequencing) in which target enrichment and PCR amplification may yield highly variable coverage profiles exome-wide, resulting in some exons being missed by chance. Whilst these areas can be discovered through bioinformatics later, re-interrogating them manually is labour intensive. Another important advantage of whole-genome sequencing is the ability to detect genome-wide structural variants (including copy number variants [CNVs]) [Citation5,Citation6]. Given the number of human disorders related to structural variants, a single test that can assess both large and small genomic variation is sometimes preferable, and the cost of whole-genome sequencing for these diseases is justified as only slightly higher than the cost of running a microarray and whole-exome sequencing separately for the same individual.
Technical considerations for study design
Because a single sequencing experiment may produce hundreds of millions of reads per sequencing lane, target coverage, and by extension variant calling quality, is highly dependent on the total number of regions being interrogated. The same number of reads that can cover a single genome for an average depth of 30× can cover a single exome (∼20 000 genes) for an average depth of >300×, representing a gross inefficiency in the use of sequencing reagents. This can be overcome using multiplexing strategies, e.g. sample barcoding, which allows sequencing more than one individual’s exome in the same sequencing lane followed by bioinformatics assignment of each read to each sample based on unique barcodes. This allows for >5 exomes to be ‘multiplexed’ in a single lane, with each being read to an average depth >60× with the same reagents consumed reading a single genome at 30× [Citation4]. This effect is multiplied several-fold as the size of the interrogated panel shrinks, e.g., >100 individuals can be investigated simultaneously for a panel of ∼200 genes in the same sequencing lane [Citation4,Citation7,Citation8]. Thus, coverage requirements and cohort size are critical variables to consider when designing NGS experiments for human disease.
NGS bioinformatics and data interpretation
One critical consideration of NGS is that instruments generate massive amounts of data, requiring sophisticated computational infrastructure and tools (bioinformatics) to process and analyse. Bioinformatics for genome sequencing is a relatively nascent field, mostly a product of work over the last decade, with algorithms and strategies adapting to rapid innovations in sequencing technologies. Regardless of the sequencing platform, all bioinformatics pipelines share in common three major aspects: read mapping, variant calling, and variant interpretation. In simple terms, read mapping is the process by which the sequenced short-reads coming off the instrument are mapped to a reference human genome by standard base-alignment methods. After mapping, bases that differ from the reference are identified (called) as variants. Once variants are called, their putative effects can be interpreted based on the genomic regions they impact and likely contribution to disease.
Variants are broadly organised into three different classes: single nucleotide variants (SNVs, previously referred to as single nucleotide polymorphisms), multi-nucleotide variants, and structural variants. Quality and zygosity of each variant are assigned based on a number of statistical considerations, including: depth of sequencing, per-base quality, the number of times each variant base is observed, and the likelihood that such a change is biologically true rather than an artefact of sequencing [Citation9]. Thus, the two steps of alignment and variant calling may themselves introduce error into the experiment, e.g. for fragments coming from highly repetitive genomic segments [Citation10]. This fact is well-recognised in the field and software development has grown into an area of intense exploration, validation, and quality guidelines [Citation11,Citation12]. Whilst some of these errors may be mitigated using long-read technologies or increasing coverage depth, these solutions remain expensive and impractical when studying large cohorts.
Perhaps the most experimentally challenging aspect of NGS bioinformatics is variant interpretation. It is at this step that the effect of each discovered variant is predicted, and thus its putative effect on disease extrapolated. Variant interpretation not only depends on a well-annotated genome, where gene and amino acid positions are well-established, but also on sequencing of large numbers of control individuals against which candidate disease variants (that are often rare) can be distinguished from population-specific polymorphisms (that may appear to be rare if inadequate numbers of population-matched controls are assessed). As more populations get sequenced around the world, these databases are expected to grow and become more robust for variant interpretation in the future [Citation13,Citation14].
NGS at the point-of-care
Nevertheless, as NGS technologies enter clinical care settings, there is a growing need to establish clinical-grade ‘gold-standard’ analysis ‘pipelines’, similar to the College of American Pathologists (CAP) or Clinical Laboratory Improvement Amendments (CLIA) certifications given to diagnostic laboratories [Citation11,Citation12,Citation15]. The role of these pipelines is to reproducibly convert a bio-specimen’s chemical signals into reproducible interpretable data, and from these data to then extract actionable information to improve patient health or change the course of disease management [Citation16]. Clearly, implementation of pipelines that can robustly cover these steps is a non-trivial task. These pipelines would need to account for several influences on quality parameters, including: sample preparation using different protocols, sequencing on different platforms, ensuring all genes in a panel are adequately covered, sources of error from the sequencing chemistry itself, and statistical errors from the sequence alignment and variant calling steps. Thus, there is a fundamental need to establish standard-operating procedures for clinical NGS that guarantee reproducibility, transparency and standardisation, thereby ensuring precision of interpretation in clinical settings.
This task scales in complexity with the number of samples being studied, and also interpretation can vary dramatically based on the population that is analysed and the databases from which annotations are being drawn [Citation17]. Of key consideration in NGS analysis is the large number of variant sites produced per individual (3–4 million per genome). Amongst these, there are hundreds or thousands of variants of unknown significance whose interpretation and relevance to health and disease is entirely unknown and can therefore neither be ruled in nor out [Citation12]. In many cases, these variants can be further stratified based on sharing with close family members, arguing for recruitment of parents and siblings at the point of care. In such cases, the presence of variants in unaffected family members may help eliminate them from further consideration; however, the converse is not true, leaving many seemingly private variants with unknown function.
Robust clinical platforms should deal with such variants accordingly, bearing in mind that some may turn up meaning in the future and may therefore be relevant to the subject’s health and should not be discarded. The fact that the field is constantly undergoing discovery, with >200 new genes and thousands of variants being linked to diseases each year in humans and many more in model organisms [Citation18,Citation19], presents a critical challenge of keeping annotation databases up to date. This has resulted in the strategy of sequencing once and interrogating often, based on the premise that a patient’s genome will not change over time and could be reassessed for causal variants periodically as annotations improve. Therefore, this strategy would support whole-genome sequencing or whole-exome sequencing methods at the point of care over targeted panels due to this potential longevity of the data. Such considerations need to be taken into account when designing clinical NGS pipelines, thus ensuring that genetic testing of patients is accurate, reproducible, and safe.
Successful application of NGS to male infertility
Infertility is defined as the inability to conceive after 1 year of continuous unprotected sexual relations [Citation20]. It affects ∼15% of couples, and males contribute to ∼50% of the causes of infertility either solely or combined with female factors [Citation21,Citation22]. There are many causes for male infertility, including genetic disorders (e.g., chromosomal anomalies or gene defects), hormonal causes, genital infection or trauma, varicocele, chemical or physical agents affecting spermatogenesis, and genital duct obstruction. Genetic anomalies have been reported in 2.2–10.8% of cases of male infertility and are higher in cases of severe quantitative infertility defects (azoospermia and severe oligozoospermia) [Citation22]. However, in 30–40% of cases of male infertility no cause can be identified and these cases are labelled ‘idiopathic’ [Citation23]. In these cases, genetic abnormalities are still highly suspected, although the genes in which they occur remain unknown.
The management of male infertility includes complete medical history taking and clinical examination followed by a combination of laboratory investigations tailored to each case. Semen analysis is the cornerstone of male infertility diagnosis. This may be followed by hormonal assays, radiological investigation and genetic studies especially in cases of severe defects. The commonly used genetic tests include karyotyping to detect numerical or structural chromosomal abnormalities and PCR to detect known genetic anomalies like Y-chromosome microdeletion, Anosmin (Kallmann syndrome) gene defects or cystic fibrosis transmembrane conductance regulator (CFTR) variants.
In the last few decades and with the advances in in vitro fertilisation (IVF) and the introduction of intracytoplasmic sperm injection (ICSI), severe male infertility cases with few sperms in semen or even cases of azoospermia with focal intra-testicular spermatogenesis can father their own children. This highlighted the need for proper genetic diagnosis to avoid vertical transmission of genetic abnormalities or production of more unstable genetic defects in the new-born. This need could be met using NGS in male infertility cohorts.
Hallmarks of disease suitability for NGS
NGS has found spectacular success in many diseases, most notably Mendelian or rare diseases, where carrying causative variants leads to significant reduction in reproductive fitness. In such cases, causative variants are rare and highly penetrant, allowing interpretation pipelines to discard the vast majority of NGS variants that are also present in control individuals or at a frequency exceeding the disease prevalence in the general population. The rarity of these variants means that other family members who are also affected are very likely to share the same genetic cause, usually due to a founder mutation that has arisen de novo in a recent common ancestor and has been maintained at low frequency in this specific family. However, one of the difficulties in NGS analysis is the issue of penetrance. Diseases where genetic variants are not completely penetrant present a daunting task for data interpretation. Similarly, situations where controls are phenotypic controls but not genetic controls (e.g., asymptomatic carriers or soon to be symptomatic carriers of late-onset diseases) require substantial complementary analysis (statistical and functional studies) to support the discovery of key causative genetic variants.
Suitability of male infertility for NGS
Male infertility is by definition a disease that significantly affects reproductive fitness, thereby ensuring causative variants remain at low-frequency in the population. However, one important difference between these variants and those that cause other rare, severe disorders is that these may be carried and passed down from females, and thus, their frequency may be higher than usually anticipated for rare diseases. Additionally, advances in IVF may lead to successful transmission of disease-causing variants if they happen to be carried in the sperm used for fertilisation. Another substantial challenge is in identifying suitable controls for research studies. Without detailed semen analysis, fertile men (with a history of fathering at least one child) should be used with caution as controls for the different types of male infertility, with the exception of azoospermia, i.e., one cannot know for sure that a confirmed father does not also suffer defects in motility, sperm morphology or sperm count.
Present systematic review
The present literature review aimed to identify the role of NGS in male infertility and to state the new genes that have been identified as a causative factor of male infertility.
In an attempt to cover all recent reports in which NGS was used to identify variants causing male infertility, we performed a search on PubMed (https://www.ncbi.nlm.nih.gov/pubmed) for combinations of the following terms: ‘exome’, ‘genome’, ‘panel’, ‘gene sequencing’, ‘whole-exome sequencing’, ‘whole-genome sequencing’, ‘next-generation sequencing’, ‘azoospermia’, ‘oligospermia’, ‘spermatogenic failure’, ‘asthenospermia’, ‘teratospermia’, ‘spermatogenesis’, and ‘male infertility’ ( [Citation24]). We restricted the search to papers published after 2010 and focusing only on humans. This process was done by a team of three individuals such that each of the papers was inspected by at least two separate individuals to determine the suitability for inclusion.
Fig. 1 Preferred reporting items for systematic reviews and meta-analyses (PRISMA) flowchart of review methodology. PubMed was searched for all articles containing NGS studies of infertility as described in the methods, limiting to the organism [Homo sapiens] and to studies published after 2010. A total of 251 unique papers were found by this search strategy, and were evaluated by at least two scientists to determine those that were germane. Studies that did not use NGS technologies, which were on female infertility, which had results from animal models, or which evaluated male patients with known developmental syndromes (e.g., PCD, Kallmann syndrome [Citation24]) were eliminated. So were studies evaluating genetics by GWAS or by single-locus interrogation (e.g., Sanger, TaqMan). Finally, studies using other omics technologies were eliminated. Altogether, 23 unique papers adhered to the inclusion criteria; all genes discovered in these studies appear in .
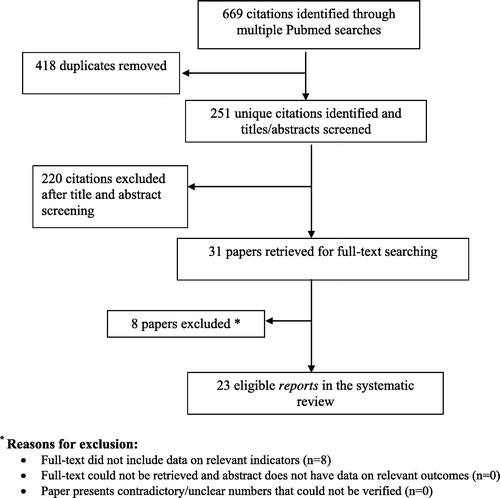
Table 1 Genetic variants discovered in infertile men by NGS technologies.
The PubMed search returned 669 articles; 418 duplicates were removed and 251 were unique. We then manually inspected all articles through the title and abstract to eliminate results that were not applicable to our present study. These included removing papers focused on: (i) female infertility; (ii) multi-organ syndromes in which infertility is a part (e.g., Kallmann syndrome, primary ciliary dyskinesia [PCD]); (iii) animal models; (iv) non-genetic characterisation of sperm or semen samples; (v) non-genetic studies using next-generation platforms (e.g., epigenetics, transcriptomics); and (vi) genetic investigation by methods other than NGS (e.g., Sanger sequencing, TaqMan, microarray, multiplex ligation-dependent probe amplification [MLPA]). In total, 220 citations were excluded after abstract screening, leaving 31 papers, which were retrieved for full-text searching. Eight papers were the excluded as the full-text did not include data on relevant indicators. Finally, 23 eligible reports were used in the present systematic review, all of which were original articles. All genes discovered in these studies are listed in [Citation25Citation[26]Citation[27]Citation[28]Citation[29]Citation[30]Citation[31]Citation[32]Citation[33]Citation[34]Citation[35]Citation[36]Citation[37]Citation[38]Citation[39]Citation[40]Citation[41]Citation[42]Citation[43]Citation[44]Citation[45]Citation[46]–Citation47].
Advances in male infertility due to NGS by subtype
Quantitative anomalies (azoospermia and oligospermia)
Perhaps the most studied of male infertility subtypes using NGS are the quantitative abnormalities: non-obstructive azoospermia (NOA) and oligospermia. The oldest of these was a study in 2013 [Citation25], in which the authors used NGS to refine a genome-wide association study (GWAS) signal they had previously discovered. In this study, five genes were interrogated around peak association signals on chromosomes 12 [peroxisomal biogenesis factor 10 (PEX10), protein arginine methyltransferase 6 (PRMT6) and SRY-box 5 (SOX5)] and 20 [signal regulatory protein α (SIRPA) and signal regulatory protein γ (SIRPG)]. Using custom-capture followed by sequencing on Illumina’s first generation Solexa platform in 96 NOA subjects and 96 healthy controls, the authors identified six variants in three genes (SIRPA, SIRPG and SOX5) that appeared at different frequencies between cases and controls [Citation25]. To verify which of these could be causal, the authors then screened only these six single nucleotide polymorphisms (SNPs) in an additional 520 NOA subjects and 477 controls. This analysis replicated only two SNVs, a protective variant in SIRPA (rs199733185) and a variant that increases risk for NOA in SIRPG (rs1048055) [Citation25]. In a separate study, Xu et al. [Citation26] also found an association between a SNV in SIRPA (rs3197744) by targeted panel sequencing of cases and controls, supporting the putative role of this gene in male infertility.
Subsequently, several other studies have used NGS to assess individuals with NOA. First, Ayhan et al. [Citation27] investigated two unrelated consanguineous families with spermatogenic failure, the first with three azoospermic brothers and one oligospermic, and the second with three azoospermic brothers. In this study, the authors used a hybrid approach of employing whole-exome sequencing after SNV genotyping, which allowed them to selectively focus on runs of homozygosity to identify the causative variant [Citation27]. This search led to the identification of a different gene for each family, TATA-box binding protein associated factor 4b (TAF4B) and zinc finger MYND-type containing 15 (ZMYND15), both harbouring recessive deleterious truncating mutations shared by all affected brothers within each family [Citation27]. Notably, the same recessive variant was shared by the oligospermic brother, suggesting some variable penetrance and supporting the grouping of quantitative abnormalities in a single category genetically.
Maor-Sagie et al. [Citation28] used whole-exome sequencing in a single patient with NOA to find a candidate homozygous splice-site mutation in synaptonemal complex central element protein 1 (SYCE1), which was then discovered to segregate with the disease in the family, i.e., one affected brother shared the same homozygous mutation, but it was absent from the fertile siblings and in heterozygous state in carrier parents, who were consanguineous. Okutman et al. [Citation29] discovered a recessive mutation in testis expressed 15, meiosis and synapsis associated (TEX15) segregating with NOA in three affected siblings in a Turkish family, absent from the fertile brother and parents. Ramasamy et al. [Citation30] discovered neuronal PAS domain protein 2 (NPAS2) mutations in three siblings with azoospermia in another consanguineous family from Turkey. Finally, Gershoni et al. [Citation31] used a combination of whole-exome sequencing and whole-genome sequencing in different families to discover mutations in the genes: meiosis specific with OB domains (MEIOB), testis expressed 14, intercellular bridge forming factor (TEX14) and dynein axonemal heavy chain 6 (DNAH6) [Citation31]. In all cases, the mutations segregated with the affected members within each family and were rare in control databases, making them prime candidates for causing disease [Citation31].
More recently, five studies published in 2017 used NGS in patients with NOA or oligospermia to uncover additional genes causative of quantitative sperm defects and male infertility. Four of these focused on multiplex consanguineous families, establishing segregation of recessive mutations in serine peptidase inhibitor, Kazal type 2 (SPINK2), MAGE family member B4 (MAGEB4), Tudor domain containing 9 (TDRD9) and adhesion G protein-coupled receptor G2 (ADGRG2) with NOA siblings but none in healthy males in the family [Citation32Citation[33]Citation[34]–Citation35]. The fifth study devised a novel experimental approach to assess both SNVs and copy number changes in 107 genes associated with male infertility from the literature [Citation36]. Using single molecular inversion probes targeting 4525 genomic regions on 21 chromosomes, the investigators were able to rapidly screen for mutations in these genes in 1138 azoospermic or oligospermic subjects [Citation36]. Whilst the authors found six infertile males with chromosomal anomalies and five with azoospermia factor (AZF)-region deletions, point mutations were only found in an additional six subjects, five with CFTR mutations and one with a mutation in synaptonemal complex protein 3 (SYCP3), further reinforcing the notion that male infertility is extremely genetically heterogeneous [Citation36]. Nevertheless, the authors comment that the sensitivity of their assay (e.g., detecting chromosomal abnormalities in patients who had already been screened by microarrays) and the cost of running such a scalable platform make it ideal for introduction into clinical settings [Citation36].
In an extension of NGS utility to the detection of structural variation, a group of 33 patients with spermatogenic failure and unexplained azoospermia were assessed by whole-genome sequencing for CNVs [Citation37]; 27 patients had a total of 42 CNVs detected, ranging in size from 40 kb to 2.38 Mb. Whilst these CNVs were distributed across multiple chromosomes, and some overlapped known CNVs common in the database of genomic variants, there were three loci that were absent from the database of genomic variants and were shared by more than one azoospermic subject: 21q22.3, 6p21.32, 13q11 each shared by two individuals [Citation37]. Only the first two of these were genic, affecting the DNA methyltransferase 3 like (DNMT3L) gene and the major histocompatibility complex, class II, DR β1 (HLA-DRB1) and major histocompatibility complex, class II, DQ α1 (HLA-DQA1) genes, respectively [Citation37]. Whilst HLA class II genes have been generally implicated in infertility [Citation48], these two genes had not been previously linked. However, evidence supporting DNMT3L gene involvement is stronger, and its role in spermatogenesis and spermatogenic impairment has been shown previously [Citation49].
Altogether, 19 genes have been implicated in causing quantitative defects in spermatogenesis by NGS technologies ().
Morphological anomalies (teratozoospermia, macrozoospermia, globozoospermia and acephalic spermatozoa syndrome)
Morphological anomalies impairing fertility occur in different forms, affecting the head, neck and the tail of the sperm. The latter usually causes motility defects (next section), whereas the former can be further subdivided into macrozoospermia, globozoospermia, acephalic spermatozoa syndrome, or dysplasia of the sperm fibrous sheath (DFS). In the era of NGS, only five studies have been published to date in which such affected subjects were sequenced. In the first of these studies, Alazami et al. [Citation38] used whole-exome sequencing in a family with asthenozoospermia, identifying a nonsense mutation in nephrocystin 4 (NPHP4). In another study, Sha et al. [Citation39] sequenced a patient with flagellar abnormalities and discovered a recessive deleterious mutation in centrosomal protein 135 (CEP135), a protein necessary for centriole biogenesis. The mutation caused infertility by forming protein aggregates in the centrosome and flagella. In a separate study, Li et al. [Citation40] discovered a mutation in bromodomain testis associated (BRDT) in a consanguineous patient with acephalic spermatozoa. The homozygous mutation, which alters a highly-conserved residue in the BRDT protein, is rare in the sense that its functional study revealed it is a gain-of-function recessive mutation [Citation40]. In this case, one suspects that the gain of function on a single allele, such as those carried by the fertile brother and father, is not sufficient to impair fertility. Moreover, in the largest study on acephalic spermatozoa syndrome, Zhu et al. [Citation41] used whole-exome sequencing in two unrelated infertile men and uncovered protein-altering recessive mutations in Sad1 and UNC84 domain containing 5 (SUN5), one individual with a homozygous variant and the other with compound heterozygous variants. This prompted Sanger sequencing of an additional 15 patients, of which six had additional recessive mutations in this gene [Citation41]. Finally, in a study of 21 patients with DFS, Sha et al. [Citation42] identified 17 unique DNAH1 mutations in 12 cases, including one homozygous and 16 compound heterozygous patients. These mutations segregated in the cases but not in unaffected family members, or a cohort of 50 ethnically matched fertile men. Using functional investigations in a subset of patients, the authors show that these subjects have diminished DNAH1 levels and disorganised 9+2 microtubule arrangements [Citation42]. Altogether, these four studies demonstrate the power of NGS in detecting causative variants in morphological sperm abnormalities.
Motility anomalies (asthenospermia and flagellar abnormalities impairing movement)
Investigation of motility anomalies using NGS has identified five unique genes from four separate studies. In the first study, Amiri-Yekta et al. [Citation43] began by investigating 10 men in six highly consanguineous families with flagellar abnormalities using whole-exome sequencing. Mutations in DNAH1 were identified in two families, and confirmed in one additional sibling from each affected family by Sanger sequencing [Citation43]. Subsequently, the authors screened an additional 38 men for the same founder mutation, identifying one more patient who shared this same mutation [Citation43]. More recently, Wang et al. [Citation44] used whole exome-sequencing to identify an additional four consanguineous Chinese men with frameshift truncating mutations in DNAH1, further establishing this gene’s role in flagellar development and motility during spermatogenesis. Further, Xu et al. [Citation45] identified homozygous mutations in two siblings of consanguineous parents with mutations affecting a highly conserved residue in sperm-associated antigen 17 (SPAG17) causing asthenospermia. Functional studies showed this mutation causes significantly decreased SPAG17 expression in the patients’ spermatozoa, consistent with a functional role in motility [Citation45].
Tang et al. [Citation46] subsequently investigated 30 independent cases with motility defects due to flagellar abnormalities and identified additional recessive mutations (homozygous and compound heterozygous) in the three cilia- and flagella-associated protein (CFAP) genes, CFAP43, CFAP44 and CFAP65 in five men. Subsequent engineering of knockout mice for two of these genes (in CFAP 43 and 44) using CRISPR (Clustered Regularly Interspaced Short Palindromic Repeats), resulted in motility and flagellar abnormalities similar to those seen in the human patients [Citation46].
Congenital bilateral absence of the vas deferens (CBAVD) and Y-chromosome NGS studies
Whilst CBAVD is usually caused by CFTR mutations, one recent study discovered mutations in the X-linked adhesion protein ADGRG2 [Citation50]. By sequencing the exomes of 12 CFTR-negative men, followed by re-sequencing the ADGRG2 gene in 14 additional men with CBAVD, they discovered four hemizygous mutations all predicted to truncate ADGRG2 [Citation50]. This is consistent with mouse studies in which male ADGRG2 knockouts develop obstruction and therefore infertility [Citation50]. A study by Oud et al. [Citation36] discovered a patient with unilateral absence of vas deferens with a CFTR mutation, further expanding the phenotyping spectrum of cystic fibrosis transmembrane receptor-based obstructive infertility.
One of the major advantages of whole-genome sequencing is the ability to detect both small and large variants, including structural and CNVs. Such approaches have been used recently on the Y-chromosome to achieve break-point resolution for CNVs [Citation51,Citation52], although no new causative genes have been identified to date. The ultra-repetitive nature of the Y-chromosome, which is rich in repeated elements and segmental duplications [Citation53], makes CNV detection challenging using whole-genome sequencing data, in particular in terms of accurate mapping of short reads. This mapping uncertainty has the potential to create false calls along the Y-chromosome, an issue that could be mitigated with long-read technologies; however, those are currently expensive and therefore not suitable for routine implementation.
Thus, given the current challenges of CNV assignment, it is no surprise that most NGS studies altogether ignore the Y-chromosome [Citation54]. Whilst recent efforts have begun to patch together Y-chromosome structural rearrangements using NGS, there have been no studies targeting infertile men to date. This represents an interesting opportunity for future investigation, further justifying the use of whole-genome sequencing for patient assessment instead of whole-exome sequencing or panel sequencing where possible.
Altogether, 23 studies have appeared to date using NGS to discover mutations in 28 genes causing a wide variety of male infertility. This number likely represents the proverbial ‘tip of the iceberg’, with >400 genes identified to cause spermatogenic impairment in mice [Citation55Citation[56]–Citation57] and up to 100 genes identified in humans in the pre-NGS era (reviewed in [Citation29,Citation58Citation[59]–Citation60]). However, as the technology is adopted more readily in clinical and research centres, it has the potential to discover many more.
Conclusions
The development and deployment of NGS technologies have the potential to transform clinical testing across a wide range of human conditions, and male infertility is clearly no exception. The work done to date is testament that whilst the investigation of infertility by modern sequencing technologies may have only recently started, it is a fantastic field to invest in from a discovery point of view. Incumbent upon the success of NGS are improvements to bioinformatics algorithms and tools that help transform data into actionable knowledge. Current test offerings are advancing from small gene panels to complete genomes, and with these advances comes an increasing need for improved bioinformatics, including analytics, annotations, and robust workflows to deliver this information to a clinical audience.
The work we review here focuses entirely on the use of NGS to uncover genetic variants in male infertility; however, NGS has now been adapted to uses outside of genomic investigations, including for example transcriptomics, epigenetics, and investigations of the microbiome [reviewed in [Citation61Citation[62]Citation[63]–Citation64]. Whilst such efforts have already begun addressing problems pertinent to male infertility (e.g., sperm cell transcriptomics [Citation65]), single-sperm cell genotyping [Citation66], spermatocyte methylation analysis [Citation67], seminal microbiome profiling [Citation68], these efforts have not reached mainstream analysis of large cohorts of affected patients. In addition to NGS-based approaches, work on spermatogenesis is flourishing with the use of metabolomics and proteomics. Detection of protein modifications, including important histone modifications such as phosphorylation, ubiquitination, sumoylation or acetylation, can shed light on gene expression patterns with functional consequences on normal (and by extension, abnormal) fertility. Similarly, studies investigating non-coding RNAs and microRNAs regulating spermatogenesis have been undertaken in males with or without infertility to discover biomarkers predictive of infertility [Citation69,Citation70]. Thus, there is substantial room to harness NGS technologies towards conceptual advances in this condition.
One of the major open questions is how can NGS be beneficial to patients with infertility, especially considering the difficulty correcting germline mutations in already affected individuals. First, we think that for many individuals, receiving a genetic diagnosis is far more meaningful than living with the ‘idiopathic’ label. The former can lead to transforming the clinical discussion from focusing on what is wrong to where to go next, rather than living a stressful, drawn-out trial and error approach of implementing various remedies in the hope of conception. Second, the availability of a diagnostic mutation could illuminate a therapeutic pathway for partially restoring fertility. Whilst the field is still in its early days with regard to genetic studies, the emerging picture of high levels of genetic heterogeneity make it well-suited for stratification of patient populations into different potential therapy groups based on affected genes and pathways. Separately, studies of these pathways may shed light on novel intervention possibilities, or opportunities to repurpose medications to improve fertility outcomes. At the very least, knowledge of the genetic mutation can be used during IVF and ICSI to select sperm cells not carrying the same mutation for male progeny.
The next decade has the potential to be defining for male infertility in particular and human diseases in general, with advances in NGS promising to play a large part. For infertile patients, there will be a long road ahead from sample collection to deriving clinical utility; in many cases, due to the significant genetic heterogeneity, the utility from any given sample will not be evident until many years down the road, when other patients with insults in the same genetic pathways are discovered. Nevertheless, patient populations should be encouraged to participate in genetic research so that those goals may one day be achieved.
Source of funding
None.
Conflict of interest
None.
Acknowledgements
These studies were supported, in part, by the Weill Cornell Medical College in Qatar; and QNRF NPRP 09-741-3-193 and NPRP 5-436-3-116.
Notes
Peer review under responsibility of Arab Association of Urology.
1 Abbreviations: ADGRG2, adhesion G protein-coupled receptor G2; BRDT, bromodomain testis associated; CBAVD, congenital bilateral absence of the vas deferens; CEP135, centrosomal protein 135; CFAP, cilia- and flagella-associated protein; CFTR, cystic fibrosis transmembrane conductance regulator; CNV, copy number variant; DFS, dysplasia of the sperm fibrous sheath; DNAH(1)(6), dynein axonemal heavy chain (1) (6); DNMT3L, DNA methyltransferase 3 like; GWAS, genome-wide association study; HLA(-DRB1) (-DQA1), major histocompatibility complex, class II, (-DR β1) (-DQ α1); ICSI, intracytoplasmic sperm injection; IVF, in vitro fertilisation; MAGEB4, MAGE family member B4; MEIOB, meiosis specific with OB domains; NGS, next-generation sequencing; NOA, non-obstructive azoospermia; NPAS2, neuronal PAS domain protein 2; NPHP4, nephrocystin 4; PCD, primary ciliary dyskinesia; SIRPA, signal regulatory protein α; SIRPG, signal regulatory protein γ; SNV, single nucleotide variant; SOX5, SRY-box 5; SPAG17, sperm-associated antigen 17; SPINK2, serine peptidase inhibitor, Kazal type 2; SUN5, Sad1 and UNC84 domain containing 5; SYCE1, synaptonemal complex central element protein 1; SYCP3, synaptonemal complex protein 3; TAF4B, TATA-box binding protein associated factor 4b; TDRD9, Tudor domain containing 9; TEX(14)(15), testis expressed (14) (15); UTR, untranslated region; ZMYND15, zinc finger MYND-type containing 15.
References
- M.L.MetzkerSequencing technologies – the next generationNat Rev Genet1120103146
- E.T.CirulliD.B.GoldsteinUncovering the roles of rare variants in common disease through whole-genome sequencingNat Rev Genet112010415425
- L.G.BieseckerW.BurkeI.KohaneS.E.PlonR.ZimmernNext-generation sequencing in the clinic: are we ready?Nat Rev Genet132012818824
- M.J.BamshadS.B.NgA.W.BighamH.K.TaborM.J.EmondD.A.Nickersonet alExome sequencing as a tool for Mendelian disease gene discoveryNat Rev Genet122011745755
- R.XiT.M.KimP.J.ParkDetecting structural variations in the human genome using next generation sequencingBrief Funct Genomics92010405415
- D.C.KoboldtD.E.LarsonK.ChenL.DingR.K.WilsonMassively parallel sequencing approaches for characterization of structural variationMethods Mol Biol8382012369384
- M.HarakalovaM.MokryB.HrdlickovaI.RenkensK.DuranH.van Roekelet alMultiplexed array-based and in-solution genomic enrichment for flexible and cost-effective targeted next-generation sequencingNat Protoc6201118701886
- M.A.QuailM.SmithD.JacksonS.LeonardT.SkellyH.P.Swerdlowet alSASI-Seq: sample assurance Spike-Ins, and highly differentiating 384 barcoding for Illumina sequencingBMC Genomics152014110
- G.A.Van der AuweraM.O.CarneiroC.HartlR.PoplinG.Del AngelA.Levy-Moonshineet alFrom FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipelineCurr Protoc Bioinformatics432013 11.0.1–33
- M.PiroozniaM.KramerJ.ParlaF.S.GoesJ.B.PotashW.R.McCombieet alValidation and assessment of variant calling pipelines for next-generation sequencingHum Genomics8201414
- A.S.GargisL.KalmanM.W.BerryD.P.BickD.P.DimmockT.Hambuchet alAssuring the quality of next-generation sequencing in clinical laboratory practiceNat Biotechnol30201210331036
- H.L.RehmS.J.BaleP.Bayrak-ToydemirJ.S.BergK.K.BrownJ.L.Deignanet alACMG clinical laboratory standards for next-generation sequencingGenet Med152013733747
- Genomes Project CG.R.AbecasisD.AltshulerA.AutonL.D.BrooksR.M.Durbinet alA map of human genome variation from population-scale sequencingNature467201010611073
- M.LekK.J.KarczewskiE.V.MinikelK.E.SamochaE.BanksT.Fennellet alAnalysis of protein-coding genetic variation in 60,706 humansNature5362016285291
- Clinical and Laboratory Standards Institute (CLSI)MM09-A2 nucleic acid sequencing methods in diagnostic laboratory medicine: approved guideline2nd ed.2014CLSIWayne, PA, USA
- S.MoorthieA.HallC.F.WrightInformatics and clinical genome sequencing: opening the black boxGenet Med152013165171
- G.H.FernaldE.CapriottiR.DaneshjouK.J.KarczewskiR.B.AltmanBioinformatics challenges for personalized medicineBioinformatics27201117411748
- J.A.KozielT.S.FranaH.AhnT.D.GlanvilleL.T.NguyenJ.H.van LeeuwenEfficacy of NH3 as a secondary barrier treatment for inactivation of Salmonella typhimurium and methicillin-resistant Staphylococcus aureus in digestate of animal carcasses: proof-of-conceptPLoS One122017e0176825
- T.AdachiK.KawamuraY.FurusawaY.NishizakiN.ImanishiS.Umeharaet alJapan's initiative on rare and undiagnosed diseases (IRUD): towards an end to the diagnostic odysseyEur J Hum Genet25201710251028
- A.MassartW.LissensH.TournayeK.StouffsGenetic causes of spermatogenic failureAsian J Androl1420124048
- I.D.SharlipJ.P.JarowA.M.BelkerL.I.LipshultzM.SigmanA.J.Thomaset alBest practice policies for male infertilityFertil Steril772002873882
- A.AgarwalA.MulgundA.HamadaM.R.ChyatteA unique view on male infertility around the globeReprod Biol Endocrinol13201537
- T.G.CooperE.NoonanS.von EckardsteinJ.AugerH.W.BakerH.M.Behreet alWorld Health Organization reference values for human semen characteristicsHum Reprod Update162010231245
- J.C.ChristianD.BixlerR.N.DexterJ.P.DonohueHypogandotropic hypogonadism with anosmia: the Kallmann syndromeBirth Defects Orig Artic Ser71971166171
- C.LuM.XuR.WangY.QinY.WangW.Wuet alPathogenic variants screening in five non-obstructive azoospermia-associated genesMol Hum Reprod202014178183
- M.XuY.QinJ.QuC.LuY.WangW.Wuet alEvaluation of five candidate genes from GWAS for association with oligozoospermia in a Han Chinese populationPLoS One82013e80374
- O.AyhanM.BalkanA.GuvenR.HazanM.AtarA.Toket alTruncating mutations in TAF4B and ZMYND15 causing recessive azoospermiaJ Med Genet512014239244
- E.Maor-SagieY.CinnamonB.YaacovA.ShaagH.GoldsmidtS.Zenvirtet alDeleterious mutation in SYCE1 is associated with non-obstructive azoospermiaJ Assist Reprod Genet322015887891
- O.OkutmanJ.MullerY.BaertM.SerdarogullariM.GultomrukA.Pitonet alExome sequencing reveals a nonsense mutation in TEX15 causing spermatogenic failure in a Turkish familyHum Mol Genet24201555815588
- R.RamasamyM.E.BakirciogluC.CengizE.KaracaJ.ScovellS.N.Jhangianiet alWhole-exome sequencing identifies novel homozygous mutation in NPAS2 in family with nonobstructive azoospermiaFertil Steril1042015286291
- M.GershoniR.HauserL.YogevO.LehaviF.AzemH.Yavetzet alA familial study of azoospermic men identifies three novel causative mutations in three new human azoospermia genesGenet Med1920179981006
- O.OkutmanJ.MullerV.SkoryJ.M.GarnierA.GaucherotY.Baertet alA no-stop mutation in MAGEB4 is a possible cause of rare X-linked azoospermia and oligozoospermia in a consanguineous Turkish familyJ Assist Reprod Genet342017683694
- M.ArafatI.Har-VardiA.HarlevE.LevitasA.ZeadnaM.Abofoul-Azabet alMutation in TDRD9 causes non-obstructive azoospermia in infertile menJ Med Genet542017633639
- Z.E.KherrafM.Christou-KentT.KaraouzeneA.Amiri-YektaG.MartinezA.S.Vargaset alSPINK2 deficiency causes infertility by inducing sperm defects in heterozygotes and azoospermia in homozygotesEMBO Mol Med9201711321149
- B.YangJ.WangW.ZhangH.PanT.LiB.Liuet alPathogenic role of ADGRG2 in CBAVD patients replicated in Chinese populationAndrology52017954957
- M.S.OudL.RamosM.K.O'BryanR.I.McLachlanO.OkutmanS.Vivilleet alValidation and application of a novel integrated genetic screening method to a cohort of 1,112 men with idiopathic azoospermia or severe oligozoospermiaHum Mutat38201715921605
- Y.DongY.PanR.WangZ.ZhangQ.XiR.Z.LiuCopy number variations in spermatogenic failure patients with chromosomal abnormalities and unexplained azoospermiaGenet Mol Res1420151604116049
- A.M.AlazamiM.J.AlshammariM.BaigM.A.SalihH.H.HassanF.S.AlkurayaNPHP4 mutation is linked to cerebello-oculo-renal syndrome and male infertilityClin Genet852014371375
- Y.W.ShaX.XuL.B.MeiP.LiZ.Y.SuX.Q.Heet alA homozygous CEP135 mutation is associated with multiple morphological abnormalities of the sperm flagella (MMAF)Gene63320174853
- L.LiY.ShaX.WangP.LiJ.WangK.Keeet alWhole-exome sequencing identified a homozygous BRDT mutation in a patient with acephalic spermatozoaOncotarget820171991419922
- F.ZhuF.WangX.YangJ.ZhangH.WuZ.Zhanget alBiallelic SUN5 mutations cause autosomal-recessive acephalic spermatozoa syndromeAm J Hum Genet992016942949
- Y.ShaX.YangL.MeiZ.JiX.WangL.Dinget alDNAH1 gene mutations and their potential association with dysplasia of the sperm fibrous sheath and infertility in the Han Chinese populationFertil Steril1071312–82017e2
- A.Amiri-YektaC.CouttonZ.E.KherrafT.KaraouzeneP.Le TannoM.H.Sanatiet alWhole-exome sequencing of familial cases of multiple morphological abnormalities of the sperm flagella (MMAF) reveals new DNAH1 mutationsHum Reprod31201628722880
- X.WangH.JinF.HanY.CuiJ.ChenC.Yanget alHomozygous DNAH1 frameshift mutation causes multiple morphological anomalies of the sperm flagella in ChineseClin Genet912017313321
- X.XuY.W.ShaL.B.MeiZ.Y.JiP.P.QiuH.Jiet alA familial study of twins with severe asthenozoospermia identified a homozygous SPAG17 mutation by whole-exome sequencingClin Genet201710.1111/cge.13059 [Epub ahead of print]
- S.TangX.WangW.LiX.YangZ.LiW.Liuet alBiallelic mutations in CFAP43 and CFAP44 cause male infertility with multiple morphological abnormalities of the sperm flagellaAm J Hum Genet1002017854864
- J.T.den DunnenS.E.AntonarakisMutation nomenclature extensions and suggestions to describe complex mutations: a discussionHum Mutat152000712
- K.van der VenR.FimmersG.EngelsH.van der VenD.KrebsEvidence for major histocompatibility complex-mediated effects on spermatogenesis in humansHum Reprod152000189196
- J.X.HuangM.B.ScottX.Y.PuA.Zhou-CunAssociation between single-nucleotide polymorphisms of DNMT3L and infertility with azoospermia in Chinese menReprod Biomed Online2420126671
- O.PatatA.PaginA.SiegfriedV.MitchellN.ChassaingS.Fagueret alTruncating mutations in the adhesion G protein-coupled receptor G2 gene ADGRG2 cause an X-linked congenital bilateral absence of vas deferensAm J Hum Genet.992016437442
- J.R.EspinosaQ.AyubY.ChenY.XueC.Tyler-SmithStructural variation on the human Y chromosome from population-scale resequencingCroat Med J562015194207
- G.D.PoznikY.XueF.L.MendezT.F.WillemsA.MassaiaM.A.Wilson Sayreset alPunctuated bursts in human male demography inferred from 1,244 worldwide Y-chromosome sequencesNat Genet482016593599
- H.SkaletskyT.Kuroda-KawaguchiP.J.MinxH.S.CordumL.HillierL.G.Brownet alThe male-specific region of the human Y chromosome is a mosaic of discrete sequence classesNature4232003825837
- A.MassaiaY.XueHuman Y chromosome copy number variation in the next generation sequencing era and beyondHum Genet1362017591603
- H.J.CookeP.T.SaundersMouse models of male infertilityNat Rev Genet32002790801
- A.N.YatsenkoN.IwamoriT.IwamoriM.M.MatzukThe power of mouse genetics to study spermatogenesisJ Androl3120103444
- D.JamsaiM.K.O'BryanMouse models in male fertility researchAsian J Androl132011139151
- M.M.MatzukD.J.LambThe biology of infertility: research advances and clinical challengesNat Med14200811971213
- G.BrebionR.A.BressanL.S.PilowskyA.S.DavidProcessing speed and working memory span: their differential role in superficial and deep memory processes in schizophreniaJ Int Neuropsychol Soc172011485493
- Y.N.LinM.M.MatzukGenetics of male fertilityMethods Mol Biol115420142537
- L.DasS.ParbinN.PradhanC.KausarS.K.PatraEpigenetics of reproductive infertility. Front Biosci (Schol Ed)92017509535
- A.SinhaV.SinghS.YadavMulti-omics and male infertility: status, integration and future prospectsFront Biosci (Schol Ed)92017375394
- L.StuppiaM.FranzagoP.BalleriniV.GattaI.AntonucciEpigenetics and male reproduction: the consequences of paternal lifestyle on fertility, embryo development, and children lifetime healthClin Epigenetics72015120
- D.T.CarrellK.I.AstonR.OlivaB.R.EmeryC.J.De JongeThe “omics” of human male infertility: integrating big data in a systems biology approachCell Tissue Res3632016295312
- Y.HongC.WangZ.FuH.LiangS.ZhangM.Luet alSystematic characterization of seminal plasma piRNAs as molecular biomarkers for male infertilitySci Rep6201624229
- Y.ShaY.ShaZ.JiL.DingQ.ZhangH.Ouyanget alComprehensive genome profiling of single sperm cells by multiple annealing and looping-based amplification cycles and next-generation sequencing from carriers of Robertsonian translocationAnn Hum Genet8120179197
- Y.DuM.LiJ.ChenY.DuanX.WangY.Qiuet alPromoter targeted bisulfite sequencing reveals DNA methylation profiles associated with low sperm motility in asthenozoospermiaHum Reprod3120162433
- S.L.WengC.M.ChiuF.M.LinW.C.HuangC.LiangT.Yanget alBacterial communities in semen from men of infertile couples: metagenomic sequencing reveals relationships of seminal microbiota to semen qualityPLoS One92014e110152
- M.JodarS.SelvarajuE.SendlerM.P.DiamondS.A.KrawetzMedicine N.ReproductiveThe presence, role and clinical use of spermatozoal RNAsHum Reprod Update192013604624
- Q.YangJ.HuaL.WangB.XuH.ZhangN.Yeet alMicroRNA and piRNA profiles in normal human testis detected by next generation sequencingPLoS One82013e66809