
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
A series of molecules containing sulfonyl and amide coupling structure were developed, synthesized and evaluated. Total 21 compounds having sulfonamide and amide groups are synthesized. The structures of the synthesized compounds were elucidated and confirmed by 1H NMR, 13C NMR, Mass spectrum and the purity was checked through HPLC analysis. All synthesized compounds (4a–4u) were tested for their in vitro anticancer activity against a series of different cell lines like A549 (Lung Cancer cell), HeLa (Cervical), MCF-7 (Breast Cancer cell) and Du-145 (Prostate Cancer cell) respectively. The results of the anticancer activity revealed that most of the tested compounds showed moderate to good anticancer activity. Compounds 4d, 4k and 4s show promising anticancer activity in different cell lines.
Introduction
The fused ring nucleus having sulfonamide and amide coupling is an important constituent for an enormous variety of therapeutic agents, including anticancer, antiproliferative, antimalarial, antifungal and antibacterial agents [Citation1Citation[2]Citation[3]Citation[4]–Citation5] . The sulphonamide act as matrix metalloproteinase inhibitors it is a significant pharmacophore and its coupling with other rings could provide new biologically active compounds [Citation6]. Lately, the applications of amide coupled with naphthoyl rings were found to show antimicrobial agents and biofilm inhibitors [Citation7]. The compounds like some esters and amide coupled compounds act as anti-inflammatory drugs as cyclooxygenase-2-inhibitors [Citation8] while some act as antimalarials [Citation9]. The sulfonamide coupled with pyrimidine shows antimicrobial and anticancer activities [Citation10]. Inhibitors of 5-Lipoxygenase [Citation11], Yersinia enteroclitica Y opH tyrosine phosphatase inhibitors [Citation12], antimalassezia [Citation13], antiamoebic and antimalarial activities [Citation14], inhibitor of phosphodiesterase type 4 [Citation15], antihypertensive [Citation16]. Some sulfonamides linked compounds act as hepatitis-C virus as nonstructural protein 3 protease inhibitors [Citation17].
The sulfonamide coupled with thiourea shows antiinflammatory and antimicrobeal activities [Citation18]. Some dihydropyrazole sulfonamide derivatives act as potential COX-1/COX-2 inhibitors [Citation19]. Literature revealed some chromone-based sulfonamide derivatives shows carbonic anhydrase inhibition and cytotoxic activity [Citation20], while the heterocyclic sulfonamides act as sphingosine 1-phosphate receptor 1 (S1P1) antagonists [Citation21].
Following a wide literature exploration, it was observed that, different coupling of sulfonamide and amide compounds shows different activities like carbonic anhydrase inhibitors [Citation22], Anti-mycobacterial [Citation23], some sulfonamides act as sphingosine-1-phosphate (S1P1) receptor [Citation24]. 11-beta-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitors for the treatment of metabolic disorders [Citation25], sulfonamide-1,2,4-triazoles, 1,3,4-thiadiazoles and 1,3,4-oxadiazoles, as potential antibacterial and antifungal agents [Citation26]. The structurally related compounds having sulfonamide and amide linkage in combinations derivatives show selectively SIRT-2 inhibiting activity [Citation27].
From above references it is clear that sulfonamide and amide coupled with different group compounds show considerable varied activities. All above references indicate that the probability of potent anticancer activity of the derivatives containing benzene sulfonamide and phenyl amide coupled compound increases considerably. So we have synthesized compounds having benzene sulfonamide and phenyl amide compounds which coupled at meta positions of benzene to each other. We have also developed simplified reaction conditions for all the steps so we can avoid costly reagents, tedious purifications, and all the synthesized compounds also have good purity. We here report the synthesis of new substituted sulfonamide derivatives () with the aim of investigating their anticancer activity. The synthetic methods adopted for the preparation of the title compounds (4a-4u) [Citation28] are depicted in the scheme presented below.
Scheme 1 Synthesis of [N-(Substituted phenyl)-2-(3-substituted) sulfamoyl) phenyl)] acetamide derivatives (4a-4u).
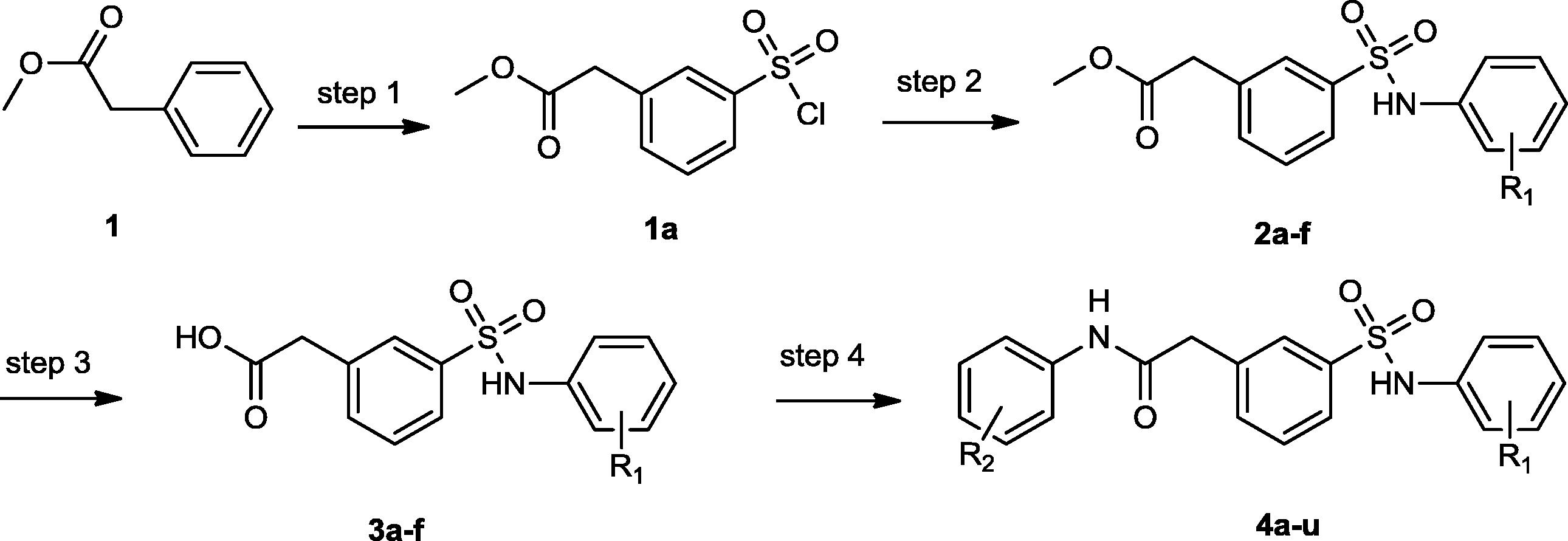
Reagents and conditions: (step 1) Chlorosulfonic acid, DCM 0 °C-rt; (step 2) substituted amine, pyridine, DCM, 0 °C -rt; (step 3) Li(OH), THF, EtOH, H2O, rt; (step 4) substituted amine, EDCI, DIPEA, DCM, rt.
From above (, Table 1 & 2 See supporting information) here we have optimized the condition for aromatic chlorosulfonation in the presence of ester group. The reactivity changes according to the equivalent of chlorosulfonic acid used. We have carried out 10 different combinations and optimized the reaction condition which reduces the efforts of tedious work up and purifications of intermediate for first time. For all the reactions we have kept time constant. It is confirmed that when we use neat excess of chlorosulfonic acid without solvent there is 60% formation of required product (entry 10), then we have used excess chlorosulfonic acid with DCM then yield is 40% (entry 9). From above these two conditions it is clear that we have to use chlorosulfonic acid in equivalents along with in neat and in DCM solvent conditions.
The varied results are shown in . The (entries 1, 2, 3 and 4) shows there is formation product along with side products, the yields are 30%–55%. When we consider (entries 5, 6, 7 and 8) the yields are increasing from 40% to 80% when we used equivalent amount of chlorosulfonic acid. Mainly there is formation of product and less side products in (entries 5–8). But in (entries 1–4) there is formation of multiple spots on TLC, but in entries 5 to 8 the TLC profile was much more promising. The yields are isolated yields after series of reactions optimization and the condition 8 works (entry 8) well for given compound. By using this method the work up is easy we have to evaporate reaction mixture under reduced pressure and obtained gummy material which is washed with excess of hexane and it is crystallized from 20% ethyl acetate: hexane mixture to obtain white solid which is used further for sulfonamide coupling reaction. In entries 1 to 4 the side product is 4-substituted sulfonyl chloride compound obtained along with polar junk material, which required purification by column chromatography so the yields are less, but in latter case purification not required pure compound obtained by crystallization which is not possible in earlier entries, the gummy material remains as such.
Table 1 In vitro anticancer screening of the synthesized compounds against four cell lines. Data are expressed as IC50 (µM) SD (n = 3).
In Scheme 2 and Scheme 3 first we have optimized the reaction solvent and base, from initial screening we have finalized DCM as the solvent and pyridine as the base that we have tabulated in Table 2. From Table 2 it is confirmed that when we used equivalents of pyridine and DCM the yield is 90% Table 4 entries 5.
There are many reports for the formations of sulfonamide so initially we have screened different bases by taking DCM and THF as solvents. In entry 5 with 2.5 equiv. of pyridine and in DCM the yield is 70% from entries 1 to 4 the yield ranges from 30% to 60%, in entries 6–10 the yield is 25%–60% also the reaction time for all entries is from 6 to 16 h. Time monitored on the bases on consumption of starting material. We have faced isolation problem in all the cases, like extraction needed for all the examples and obtained compounds are not cleaner so need to modify the yields. In entry 5 we have optimize work up condition so that we have to avoid rigorous extractions. In Table 3 we have varied the equivalents of pyridine and we have come to conclude that if we use equivalent volumes of pyridine along with DCM solvent then yield is 90% (entry 5). We have used pyridine as base and modified the work up by treating the reaction mass with cold 2N aq. HCl and stirred reaction mass for 30 min., the solid precipitates out in all the intermediates which is filtered and washed it with cold diethyl ether and cold pentane, all the intermediates obtained are white solids. Most of examples the yield of solid is 70% to 80% further we have extracted the remaining aqueous layer with DCM and evaporated it to obtained the remaining solids ultimately yield increases up to 80% to 90% in all the intermediates.
For Scheme 4 we have used excess of lithium hydroxide 5 equiv. we have modified the reaction work up condition, by giving back wash in basic condition and later acidifying it to get desired product with required purity and no column purification is required all the acids obtained are enough pure to used further for next amide coupling with different amines. All intermediates gives yield up to 80% to 85% in most of cased and all the compounds obtain as white solids.
Scheme 5 we have screened different coupling reagents, bases and solvents it is concluded that when we use EDCI (1.5 equiv) along with DIPEA (2.5 equiv) in DCM the yields is 90% last entry. We have varied different coupling reagents, different bases and different reaction time in DMF and DCM (Table 4). We have done series of peptide coupling reactions using HATU, along with triethyl amine and DIPEA the yields are in between 50% and 55%. With PyBoP used triethyl amine and DIPEA again yields are in 47%–50%. With most commonly conditions EDCI and HOBt using triethyl amine and DIPEA in DCM and DMF solvents the yields are in the range of 50%–70%.
In all above examples the aqueous work up required, the reagents are costly and in all the cases the yield obtained after column purifications. But, the condition in entry 10, requires the extraction, and later on washing with 2N aq. HCl washing to obtain 70% to 80% pure product which was washed with 5% DCM and Hexane, cold diethyl ether and cold pentane gives the desired compounds with 95% and above purity and yield is also 90% in most of synthesized compounds. We have optimized all the 4 steps, no need of column chromatography, no costly reagents required, no prep purification required. All obtained compounds are with 95% and above purity.
From above data we decided the synthesis of [N-(Substituted phenyl)-2-(3-substituted) sulfamoyl) phenyl)] acetamide derivatives (4a-4u) ().
The synthesized compounds were evaluated for their in vitro anticancer activity against human lung cancer cell line (A549), cervical (HeLa) cancer cell line, breast cancer cell line (MCF-7) and prostate cell line (DU-145) using 5-fluorouracil as reference drug [Citation29]. The response parameter calculated was the IC50 value, which corresponds to the concentration required for 50% inhibition of cell viability. The results are presented in , where all compounds exhibit moderate to good activity compared to 5-fluorouracil as positive control.
In the case of the human lung cancer cell line (A549) compounds 4a, 4b, 4d, 4k and 4s were the most potent, with IC50 values ranging from 1.81 to 2.11 µM. On the HeLa cell line the compounds which showed potent activity were 4b, 4d, 4k, 4n, 4p and 4s (IC50 = 1.92–2.52 µM, respectively). In case of the MCF-7 breast cancer cell line, the potent compounds were 4b, 4d, 4k and 4s. (IC50 = 2.12–2.52 µM). Moderate activity was observed for the synthesized compounds on the Du-145 prostate cancer cell line, where the most potent candidates were compounds 4k, 4p and 4s (IC50 = 2.12–2.76 µM). Generally, the lung (A549) and cervical (HeLa) cancer cell lines were the most sensitive to the synthesized compounds.
With regard to broad spectrum anticancer activity, close examination of the data presented in , reveals that compounds 4d, 4k and 4s were the most active, showing effectiveness toward the four cell lines.
The SAR can be explained on the basis of substitutions on both the aromatic rings less hindered substitution like methyl and ethyl on ortho and para position of rings increases the anticancer activity in all four cell lines, interestingly ortho triflouromethyl and indoline group decreases the anticancer activity and despite steric hindrance 4d, 4k and 4s shows promising activity because electron donating tendency. Most of the compounds show promising anticancer activity with electron donating groups on the ring than electron withdrawing groups.
Conclusion
In conclusion by using this methodology a series of molecules containing sulfonyl and amide coupling structures are synthesized and evaluated. We have developed easy and effortless method for the synthesis of some sulfonyl and amide coupling derivatives by simple reaction steps. The method is economical in the sense that no expensive reagents are required, no any tedious purification is needed and all the compounds synthesized are obtained in good yields. The advantages of this method are mild reaction conditions, shorter reaction time and potential anticancer activity. The compounds (4d, 4k and 4s) show potent anticancer activity in all the four cell lines tested.
Supplementary data
Download MS Word (8.7 MB)Acknowledgements
The authors are thankful to the Head, Department of Chemical Technology, Dr. Babasaheb Ambedkar Marathwada University, Aurangabad 431004, MS, India for providing the laboratory facility.
References
- K.P.BhabakC.ArenzNovel amide- and sulfonamide-based aromatic ethanolamines: effects of various substituents on the inhibition of acid and neutral ceramidasesBioorg Med Chem20201261626170
- M.T.TavaresK.F.M.PasqualotoJ.V.D.Streeket al.Synthesis, characterization, in silico approach and in vitro antiproliferative activity of RPF151, a benzodioxole sulfonamide analogue designed from capsaicin scaffoldJ Mol Struct10882015138146
- R.PingaewV.PrachayasittikulA.Worachartcheewanet al.Novel 1,4-naphthoquinone-based sulfonamides: synthesis, QSAR, anticancer and antimalarial studiesEur J Med Chem1032015446459
- M.SunH.H.YangL.Tianet al.Design, synthesis, and fungicidal activities of imino diacid analogs of valine amide fungicidesBioorg Med Chem Lett25201557295731
- M.KumarB.NarasimhanP.Kumaret al.4-(1-Aryl-5-chloro-2-oxo-1,2-dihydro-indol-3-ylideneamino)-N-substituted benzene sulfonamides: Synthesis, antimicrobial, anticancer evaluation and QSAR studiesAra J Chem72014436447
- P.JainC.SaravananS.K.SinghSulphonamides: deserving class as MMP inhibitors?Eur J Med Chem60201389100
- S.NizalapurK.K.K.HoO.Kimyonet al.Synthesis and biological evaluation of N-naphthoyl-phenylglyoxamide-based small molecular antimicrobial peptide mimics as novel antimicrobial agents and biofilm inhibitorsOrg Biomol Chem14201636233637
- A.S.KalgutkarA.B.MarnettB.C.Crewset al.Ester and amide derivatives of the nonsteroidal antiinflammatory drug, indomethacin, as selective cyclooxygenase-2 inhibitorsJ Med Chem43200028602870
- S.AnushaA.SinhaC.P.B.Rajeevet al.Synthesis, characterization and in vitro evaluation of novel enantiomerically-pure sulphonamide antimalarialsOrg Biomol Chem1320151068110690
- K.EaswaramoorthiA.J.RajendranK.C.Raoet al.Synthesis of novel 1,4-disubstituted 1,2,3-triazolo-bosentan derivatives – evaluation of antimicrobial and anticancer activities and molecular dockingRSC Adv52015105266105278
- S.SinhaT.V.SravanthiS.Yuvarajet al.2-Amino-4-aryl thiazole: a promising scaffold identified as a potent 5-LOX inhibitorRSC Adv620161927119279
- P.G.A.MartinsA.C.O.MenegattiL.D.C.Delatorreet al.Synthetic chalcones and sulfonamides as new classes of Yersinia enterocolitica YopH tyrosine phosphatase inhibitorsEur J Med Chem6420133541
- V.K.ThulamS.C.B.KotteH.S.Kumaret al.A novel and efficient method for N-acylation of sulfonamides and carbamates: their biological evaluation towards anti Malassezia activityJ Pharm Res72013195199
- A.SalahuddinA.InamR.L.Zylet al.Synthesis and evaluation of 7-chloro-4-(piperazin-1-yl)quinoline-sulfonamide as hybrid antiprotozoal agentsBioorg Med Chem21201330803089
- S.V.ReddyG.M.RaoB.V.Kumaret al.Novel imidazophenoxazine-4-sulfonamides: their synthesis and evaluation as potential inhibitors of PDE4Bioorg Med Chem21201319521963
- J.LiuJ.Q.LiuX.Yanget al.Design, synthesis, and biological evaluation of 1,2,4-triazole bearing 5-substituted biphenyl-2-sulfonamide derivatives as potential antihypertensive candidatesBioorg Med Chem21201377427751
- T.A.KirschbergN.H.SquiresH.Yanget al.Novel, sulfonamide linked inhibitors of the hepatitis C virus NS3 proteaseBioorg Med Chem Lett242014969972
- A.P.KecheV.M.KambleSynthesis and anti-inflammatory and antimicrobial activities of some novel 2-methylquinazolin-4(3H)-one derivatives bearing urea, thiourea and sulphonamide functionalitiesAra J Chem201410.1016/j.arabjc.2014.10.025
- Z.ChenZ.WangX.Yanet al.Design, synthesis, biological evaluation and molecular modeling of dihydropyrazole sulfonamide derivatives as potential COX-1/COX-2 inhibitorsBioorg Med Chem Lett25201519471951
- F.M.AwadallahA.T.El-WaeiM.M.Hannaet al.Synthesis, carbonic anhydrase inhibition and cytotoxic activity of novel chromone-based sulfonamide derivativesEur J Med Chem962015425435
- E.J.HennessyG.GrewalK.Bythet al.Discovery of heterocyclic sulfonamides as sphingosine 1-phosphate receptor 1 (S1P1) antagonistsBioorg Med Chem Lett25201520412045
- L.SyrjanenA.L.VermelhoI.A.Rodrigueset al.Cloning, characterization, and inhibition studies of a β–carbonic anhydrase from Leishmania donovani chagasi, the protozoan parasite responsible for leishmaniasisJ Med Chem56201373727381
- B.L.WilkinsonL.F.BornaghiA.D.Wrightet al.Anti-mycobacterial activity of a bis-sulfonamideBioorg Med Chem Lett17200713551357
- H.XuH.ZhangL.Luanet al.Discovery of thiadiazole amides as potent, S1P3-sparing agonists of sphingosine-1-phosphate 1 (S1P1) receptorBioorg Med Chem Lett22201224562459
- D.S.YoonS.C.WuR.Seethalaet al.Discovery of pyridyl sulfonamide 11-beta-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitors for the treatment of metabolic disordersBioorg Med Chem Lett24201450455049
- P.ZoumpoulakisC.CamoutsisG.Pairaset al.Synthesis of novel sulfonamide-1,2,4-triazoles, 1,3,4-thiadiazoles and 1,3,4-oxadiazoles, as potential antibacterial and antifungal agents. Biological evaluation and conformational analysis studiesBioorg Med Chem20201215691583
- M.A.KhanfarL.QuintiH.Wanget al.Development and characterization of 3-(benzylsulfonamido)benzamides as potent and selective SIRT2 inhibitorsEur J Med Chem762014414426
Further reading
- General experimental procedure for the synthesis of compound 4a-4u Step-1: Preparation of 2-(3-chlorosulfonylphenyl) acetate (1a) To a stirred solution of methyl 2-phenylacetate (40 g, 266 mmol) in DCM (100 mL). RM was cooled to 0 °C and Chloro sulfonic acid (34 g, 293 mmol) was added drop wise followed by stirring at room temperature for 1 h. The reaction was monitored by LCMS and TLC, after completion of reaction, evaporate reaction mixture under reduced pressure and obtained gummy material is washed with excess of hexane and it is crystalized from 20% ethyl acetate: hexane mixture to obtain white solid as 2-(3-chlorosulfonylphenyl)acetate (1a) which is used further for sulfonamide coupling reaction, Yield- (54 g, 81%). Step-2: General experimental procedure for preparation of 2a-2f To a stirred solution of 2-(3-chlorosulfonylphenyl)acetate (1) (1 equiv) in DCM (10 times) was added Pyridine (10 times) the mixture was stirred at rt for 15 min. RM was cooled to 0⁰C and substituted amine (1.5 equiv) was added drop wise followed by stirring at rt for 6 h. The reaction was monitored by TLC and LCMS, after completion of reaction poured reaction mass on cold 2N aqueous HCl and stirred it for 30 min. The precipitation formed in RM. Filtered the obtained solid and washed it with excess of water and cold diethyl ether and cold pentane to obtain all compounds as white solids. For filtrate extracted with DCM twice. The organic layer was washed with brine solution; organic layer was evaporated under reduced pressure to get desired products as white solids. But in most of cases solid compound yields are in 70-80%. Yield- 80-90%. Step-3: General experimental procedure for preparation of 3a-3f To a stirred solution of compound 3 (1 equiv) in THF (10 times) added Ethanol (4 times) and water (2 times), finally added lithium hydroxide (5 equiv) and stirred reaction mixture for 8 h. Progress reaction was monitored by TLC and LCMS. After the completion of reaction, evaporate reaction mixture under reduced pressure to obtained gummy material. Added 10 ml of water in it and extracted it with diethyl ether (10 ml). Collected aqueous layer and adjust its pH to 4 by using 6N aqueous HCl. Precipitation occurs stirred it for 30 min. Filtered the obtained solid and wash it with excess of water, cold diethyl ether (10 mL) and cold pentane (10 mL) to obtain desired compounds as white solids. Yield- 80%-90%. Step-4: General experimental procedure for preparation of 4a-4u The acid (1 equiv.) was dissolved in DCM and treated with EDCI (1.5 equiv), DIPEA (2.5 equiv). Reaction mass stirred for 10 min. then added amine (1.5 equiv) and stirred reaction mixture at room temperature for 8 h. The reaction was monitored by TLC. Added 15 ml of cold water and stirred for 20 min. Then extracted it with 20 ml of DCM .Collected organic layer wash it with 1N aqueous HCl (10 mL) and washed with brine (10 mL). Evaporate the organic layer to obtain compound with 70 to 80% purity of compounds 4a to 4z. Purification done by washing with 5:95% of DCM: hexane. The obtained solid was washed with cold diethyl ether (20 mL) and cold pentane (20 mL) to obtain compounds 4a-4u as white solids (80-90% yield).
- Biological Methods Cell Culture Human cancer cell lines HeLa (cervical), A549 (lungs) andDU-145 (prostate) were grown in DMEM + GlutaMax (Invitrogen, Carlsbad, CA, USA), and MCF-7 (breast) were grown in DMEM-F12 + GlutaMax) medium (Invitrogen), supplemented with 10% heat-inactivated bovine serum (Gibco) and penicillin-streptomycin (Gibco, Gaithersburg, MD, USA) at 37 °C in a humified chamber with 5% CO2 supply Cytotoxicity Assay Cells were seeded (105 cells/well) in 96-well flat-bottom plates (Becton-Dickinson Labware, Franklin Lakes, NJ, USA) a day before treatment and grown overnight. Compounds were dissolved in dimethyl sulfoxide (DMSO; Sigma) and finally prepared as 1.0 mg/mL stocks, respectively in the culture media. The final concentration of DMSO never exceeded 0.1% in the treatment doses. Six different doses of compounds (400, 200, 100, 50, 25 and 10 µM) were further prepared by diluting the stocks in culture media, and cells were treated (in triplicate/dose). 5-fluorouracil was included as standard reference drug (positive control) and untreated culture was considered as negative control. The cultures were further incubated for 48 hrs. At 48 h post-treatment, cell viability test was performed using TACS MTT Cell Proliferation and Viability Assay Kit (TACS) as per manufacturer’s instructions. The optical density (OD) was recorded at 570 nm in a microplate reader (ELx800, BioTek, Winooski, VT, USA) and cell survival fraction was determined. The cell survival fraction was calculated as [(A-B)/A], where A and B are the OD of untreated and of treated cells, respectively. The concentration required for 50% inhibition of cell viability (IC50) was calculated and compared with the reference drug 5-fluorouracil and the results are given in Table 1.
Supplementary data associated with this article can be found, in the online version, at https://doi.org/10.1016/j.ejbas.2017.09.001.