
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
The structural properties of Hypoxanthine compound related to molecular dynamic activity have been ornately interpreted in this methodical research work. The FT-IR, FT-Raman, NMR and UV–Visible spectral analysis has been carried out and the obtained results were validated using theoretical tools. The tautomerism of the compound has been monitored while analyzing the molecular structure regarding to describe unknown properties and applications. The displacement of chemical shift of core carbons in pyrimidine and Imidazole rings by the injection of = O atom and the mechanism customized to induce chemical properties were interpreted. The electronic degeneracy pathway in the interaction orbitals was discussed by viewing the frontier molecular overlapping. The involvement of CT-complex transitions in non bonding molecular orbitals has been inspected. The QSAR properties, Ligand efficiency (LE), Lipophilic Efficiency (LipE), were calculated and reported for explaining biological activity of Hypoxanthine. The drug and biological activity of the present compound was interpreted and the capability of suitable applications has been interpreted. The VCD spectrum for the measurement of toxicity level was simulated and the rate of masking was examined.
Introduction
The Hypoxanthine is normally belongs to heterocycles family which peculiarly contains pyrimidine and imidazole rings. Hypoxanthine is a purine derivative with nitrogenous base rarely found as a constituent nucleic acid [Citation1]. Heterocycles showing in their strange structures where the pyrimidine ring is fused to azolic moieties, are interesting systems being with important biochemical, pharmacological and physicochemical property [Citation2,Citation3] . During the recent decades numerous pyrimidine derivatives have found to have wide clinical and pharmacological applications [Citation4]. Particularly, fused pyrimidine with Imidazole ring persists to attract considerable attention since their great practical usefulness, primarily due to very wide spectrum of biological activities [Citation5,Citation6] . The hypoxanthine naturally has different Tautomerism which resolves the specific prototype of hydrogen bond donors and acceptors present in a specific molecule. Similarly, the fused imidazole and its derivatives are generally having bioactivity against inflammatory mediators [Citation7].
The organic Tautomerism molecule and mutual linked ligand H-bonds is very significant to provide the biochemical and pharmacological property [Citation8]. The natural occurring of tautomerizations is very important process which is significant for the molecule being with biochemical and pharmacological applications [Citation9,Citation10] . When Heterocycles is supposed to have heteronuclear atoms, it can be acted as an ideal ligand which is able to delocalize and thereby stimulate resonance hybridization to interact with strong intermediate to produce the sensitive antimalarial drugs [Citation11,Citation12] . Hypoxanthine is also a nutrient additive for a variety of cell culture applications by which it involves bacterial, parasite (Plasmodium falciparum) and animal cells. Hypoxanthine is an important component for the selection media used in hybridoma technologies (see ).
Table 1 Optimized geometrical parameters for Hypoxanthine computed at HF/DFT (B3LYP&B3PW91) with 6-31++G(d,p) & 6-311++G(d, p) basis sets.
After monitoring the literature, so far, no work has been found to determine the reaction path mechanism and structure activity relationship on hypoxanthine for the interpretation of physico-chemical property. In this work, the detailed analysis is carried out to find out the background reason of the compound to be pharmaceutical active. The experimental and theoretical molecular spectroscopy tools have been used for investigating the molecule.
Experimental profile
Physical state:
| • | The raw compound; Tetrachlorophthalic acid has been taken in liquid form which is pure and spectroscopic grade for acquiring spectra. | ||||
Recording profile:
| • | The FT-IR and FT-Raman spectra of the compound were recorded in Bruker IFS 66 V spectrometer and the instrument adopted with an FRA 106 Raman module equipped with a Nd:YAG laser source operating at 1.064 µm line widths with 200 mW power [Citation13]. | ||||
| • | The well distinct 1HNMR and 13CNMR spectra were recorded using high resolution 1200 MHz FT-NMR spectrometer [Citation14]. | ||||
| • | The UV–Vis spectrum was recorded in the range of 200 nm–800 nm, with the scanning interval of 0.2 nm, using the UV–1700 series instrument [Citation15]. | ||||
Computational methodology
In order to calculate the structural, spectral, Mulliken charge distribution, molecular orbital interactions and CT complex on the electronic structure parameters, the entire quantum calculations were performed using the Gaussian 09 D. 01 version, Software program in 7th Gen computer with core i7 processor [Citation16].
The entire computational process is performed using HF and DFT-B3LYP and B3PW91 methods adopted with 6-31++G(d, p) and 6-311++G(d,p) basis sets. The energy absorbance modulated electronic spectra, NBO and Frontier energy levels have been identified and validated using time-dependent SCF method with suitable basis set. In similar way, the 1H and 13C NMR chemical shifts on par with TMS were calculated by GIAO method using I-PCM model with B3LYP/6-311++G(2d,p). The dipole moment, chemical hardness, softness, electrophilicity index and chemical reactivity index were calculated from occupied and unoccupied energy levels. The linear polarizability and the first order hyper polarizability in different coordinates were computed using B3LYP method with the 6-311++G(d,p) basis set. The VCD spectra were simulated from sequential absorption and scattering frequencies and the optical chirality pattern was discussed. The biological activity parameters and QSAR properties were calculated using Molinspiration module.
Results and discussion
Structural deformation analysis
The present molecule is fabricated by fusing of Imidazole ring with pyrimidine ring and the molecular sequence in crystal unit cell was shown in . As in the figure, when the double bond existed between ring C and O can be delocalized to make double bond between C and N in the ring forms another imine group in the same ring. Thus, the resonance effect was found by forming hydroxyl group. In this venture, the double bond was found to be more polarized and the corresponding π-bond length was reduced much when compared with the literature [Citation17].
Fig. 1 molecular structure of Hypoxanthine.
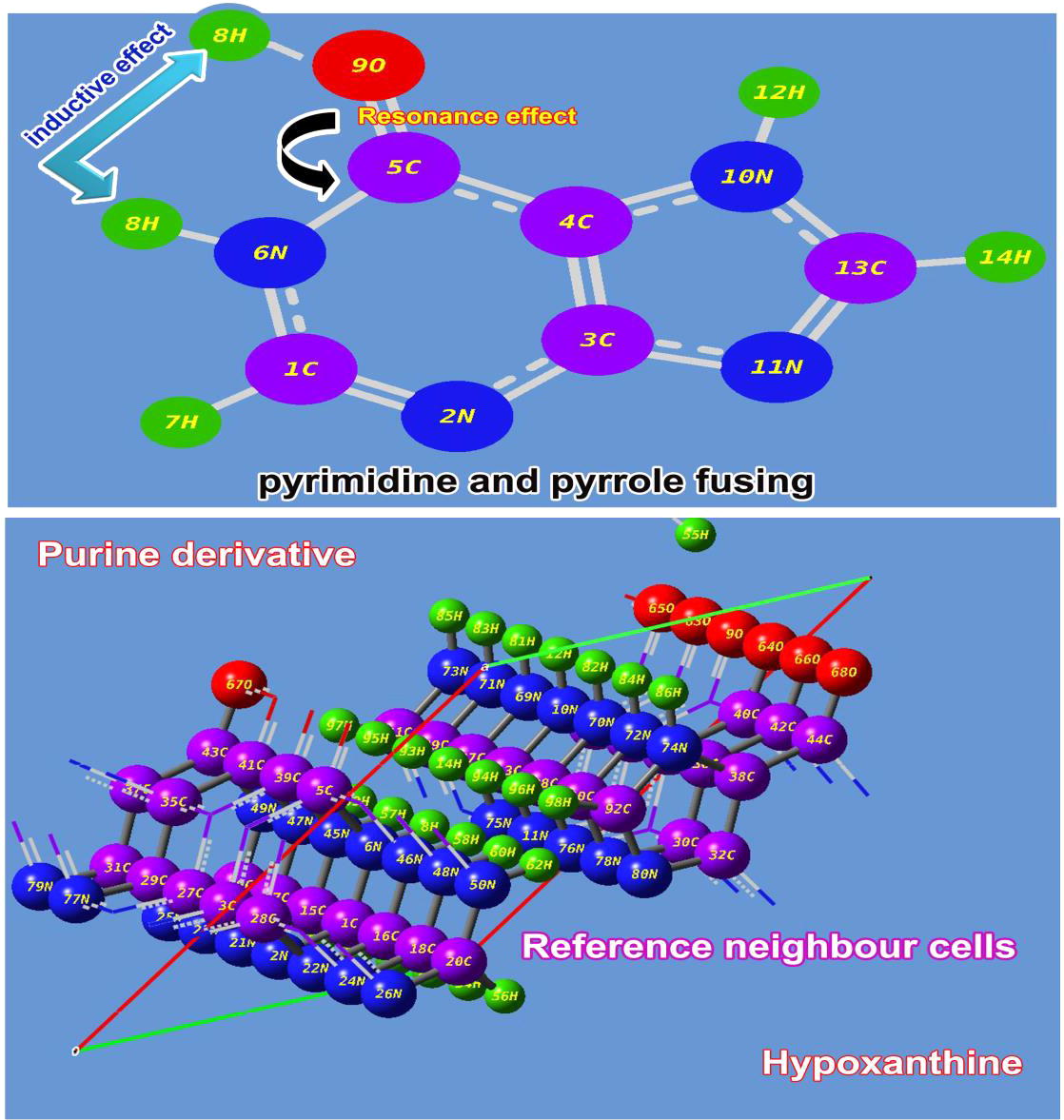
Since the existence of inductive and resonance effect in the rings and adopted atoms, the molecule is much reactive and induce new drug property. In this molecule, there were eight C-N bonds found, among them, the bond length of N11-C13 and C1-N2 were lesser than rest of others since it was imine group of bonds. The bond length of C-N in pyrimidine ring found to be lesser than imidazole ring in order to comfort the pyrimidine ring. The heteronuclear bonds C4-N10, C3-N11, C1-N6 and N2-C3 were having approximately same bond length which was less than C5-N6 which is due to the existence of =O. The arrangement of hetero nuclear atoms in the chemical bonds in the molecule is usually increase the covalent energy in considerable way which tends to produce the increment of molecular orbital energy difference. This situation in the composite molecule generates the hyper chemical activity which may be intensive pharmacological character.
Mulliken charge profile
The chemical equilibrium reinforced attractive and repulsive interactions acted on the molecular orbitals and tends to arrange local electronic charges over the atoms which are very important to display the dipole moment, strong & weak interactive bonds and electrophilic attack regions [Citation18]. The Mulliken charges orientation mainly explains the accumulation of potential energy among different entities upon which the grid electrostatic potential surfaces can be drawn [Citation19,Citation20] . The Mulliken charge displacement diagram of Hypoxanthine was displayed in the in .
Fig. 2 Mulliken charge profile of Hypoxanthine.
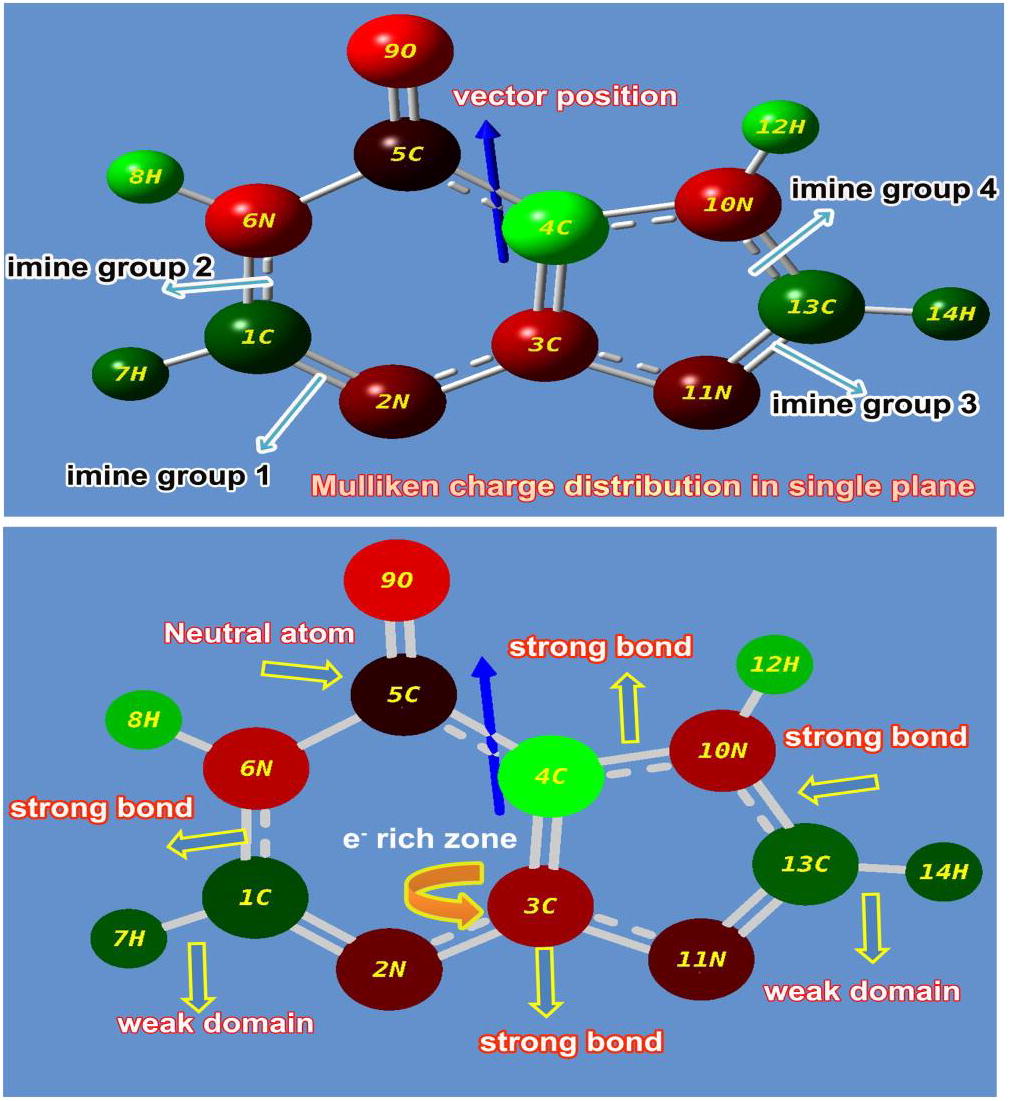
Generally, the existence of imine group in heterocyclic compounds induced the potentially beneficial activity in pharmaceutical and biological field [Citation21]. Here, four strong imine bonds have been set up among the rings and is very significant to exhibit unique properties and novel biological and antibiotic reactivity. In the pyrimidine ring, the carbonyl group was found to be formed by SP2 hybridization which enriching the drug activeness. It also found as neutral atom which was denoted by black colour and it can be considered as energy exchange point from left moiety to right moiety and further energy was transferred to Imidazole ring in order to blend the pyrimidine and Imidazole property. The electronic content were found to be very concentrated over the nitrogen atoms and proportionately, the proton content was maintained over the C of the both rings so as to preserve chemical equilibrium. In this case, the electron cloud was observed over N of rings, so proton cloud concentrated over C invariantly for the drug productivity.
Structure activity/property relationship
Generally, the physicochemical properties can be interpreted by computing SAR parameters; Surface area grid (SAG), molar volume (V), hydration energy (HE), partition coefficient octanol/water (logP), molar refractivity (MR), Polarizability (Pol) and molecular weight (MW). The results were obtained using HyperChem 8.0.6 software and depicted in and others were calculated using Molinspiration online database (TPSA and nrotb) [Citation22Citation[23]–Citation24] . The entire chemical parameters and structure activity parameters were presented in and Topographical polar surface area & lipophilicity diagrams of present compound displayed in .
Table 2 Bioactivity values of Hypoxanthine.
Fig. 3 Molecular properties diagram of Hypoxanthine.
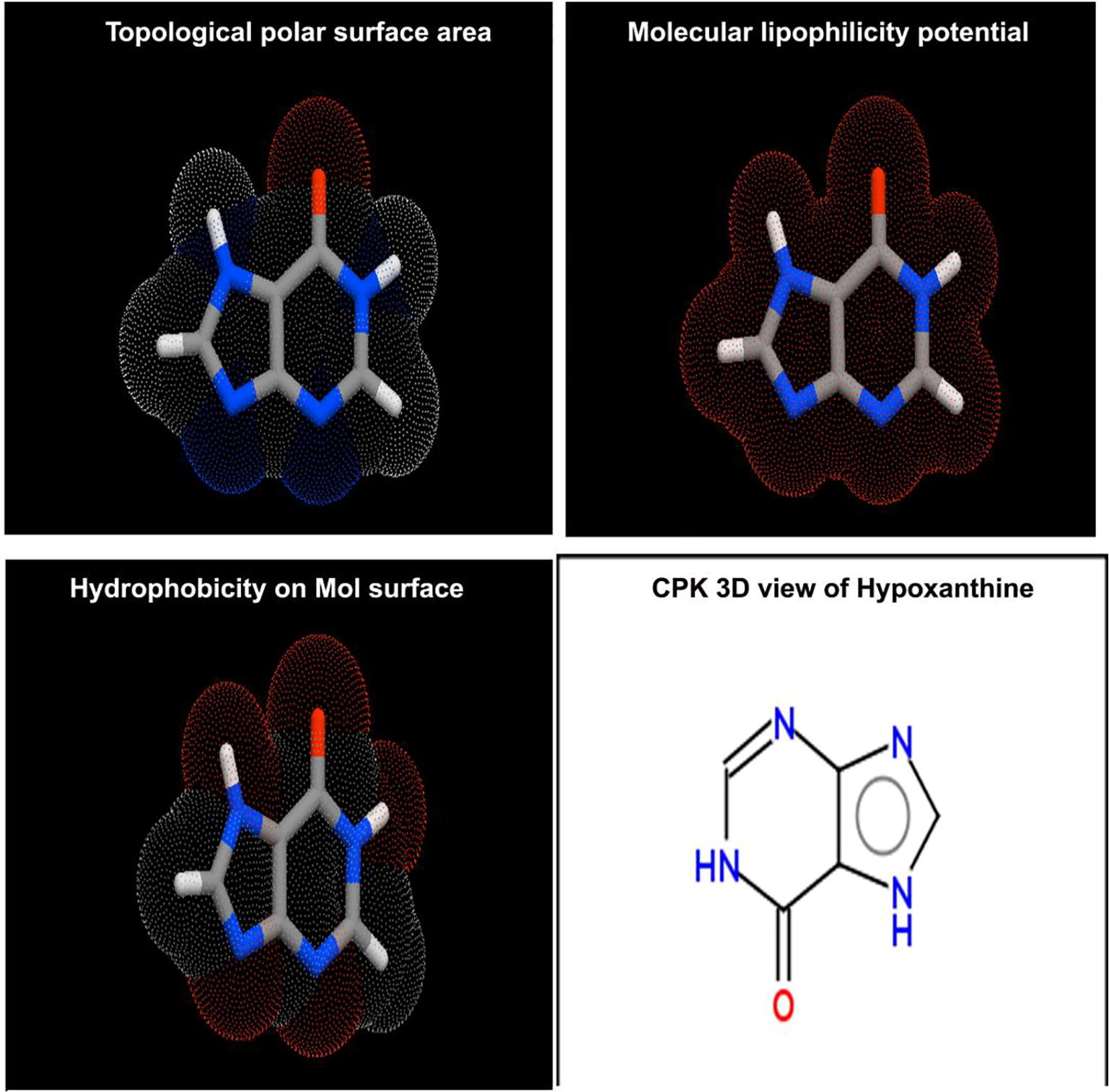
The first and foremost significant topological parameter is a number of rotatable bonds (nrotb) in a compound by which molecular flexibility can be measured and is treated to be a good narrator of oral bioactivity of drug [Citation25]. Accordingly, the nrotb was found to be nil and it should be less than 10 for the molecule being much flexible which showed the present molecule was having additional flexible. The Topological polar surface area (TPSA) of the compound is a significant to guess the capability of drug transport properties. This parameter is normally correlated with the attitude of human intestinal absorption and Caco-2 monolayer’s permeability [Citation26]. Therefore, such as was found to be 70.1 A2 which demonstrated rich intestinal absorption.
The Log P is mainly used to measure the solubility of oral drug and if it is increases to the maximum level, the solubility in water will be amplified, concurrently, the absorption will be increased. Here, the Log P was appeared to be 1.73 and was sensible. Thus, the present chemical compound was able to have good aqueous-solubility, better gastric tolerance and efficient elimination through the kidneys. If a compound is having Molecular Weight lesser than 500, it will be able to pass through cell membrane. Here, the value was found to be 136.11 g/mol, is capable to penetrate the cell membrane. The present molecule has H-bond donor and acceptor count 2 respectively and hence the present drug able to penetrate the cell and this accepted the Lipinski’s limit by which the compound displayed its druglikeness [Citation27]. The ligand efficiency was identified as 4 and the lipophilicity of the molecule was found to be 11. These are the imperative aspect in which the present drug possesses high-quality oral bioavailability. The rotatable ligand bond count is very important to describe the complexity and enantiomer affecting factor which was 0 for the present compound and it can be as simple and less toxicity molecule.
The heavy atom count of the aromatic molecule is usually making dynamic chemical potential. Accordingly, the heavy atom count was found to be 10 which is very high and molecule was having potential energy for producing consistent drug activity. The GPCR is G protein–coupled receptors (GPCRs) which also known as seven-transmembrane domain receptors and was found to be 0.80 for the present case. Here, the value showed the good signal transduction. The Ion channel modulator value was determined as 0.64 which is enough to modulate the Ion channels are pore-forming membrane proteins that allow ions to pass through the channel pore. The kinase inhibitor of present case was 0.58 which was enough to modulate its function of protein kinases. The nuclear receptors are multifunctional protein which play key roles in both embryonic development and adult homeostasis which was found to be 3.56 and it was able to transduce signals of their cognate ligands. The Protase inhibitor is an antiviral character of the compound and here the same was found to be 1.97 and it was showed that, the present compound will be acting as antiviral drug.
Vibrational analysis
The vibrational analysis is significant to determine the vibrational region of compositional bands through which the compositional parts of the compound can be identified. Here, the credible fundamental vibrational pattern of Hypoxanthine was recognized by IR pattern and obsessed wavenumbers was presented in . The scanned FT-IR and FT-Raman vibrational wavenumbers of recorded and simulated spectra (HF and DFT) were exposed in the Figs. 4 and 5 respectively. The actual molecule was tailored by pyrimidine ring fused with Imidazole ring. The resultant compound consists of 14 entities and the structure belongs to CS point group. The 36 fundamental modes of vibrations were dispersed as 14 modes were identified as stretching, 11 modes were represented by in plane bending and 11 peaks recognized as out of plane bending vibrations.
Table 3 Observed and HF and DFT (B3LYP & B3PW91) with 6-31++G(d,p) & 6-311++G (d,p) level calculated vibrational frequencies of Hypoxathine.
Fig. 4 Experimental and Calculated FTIR spectra of Hypoxanthine.
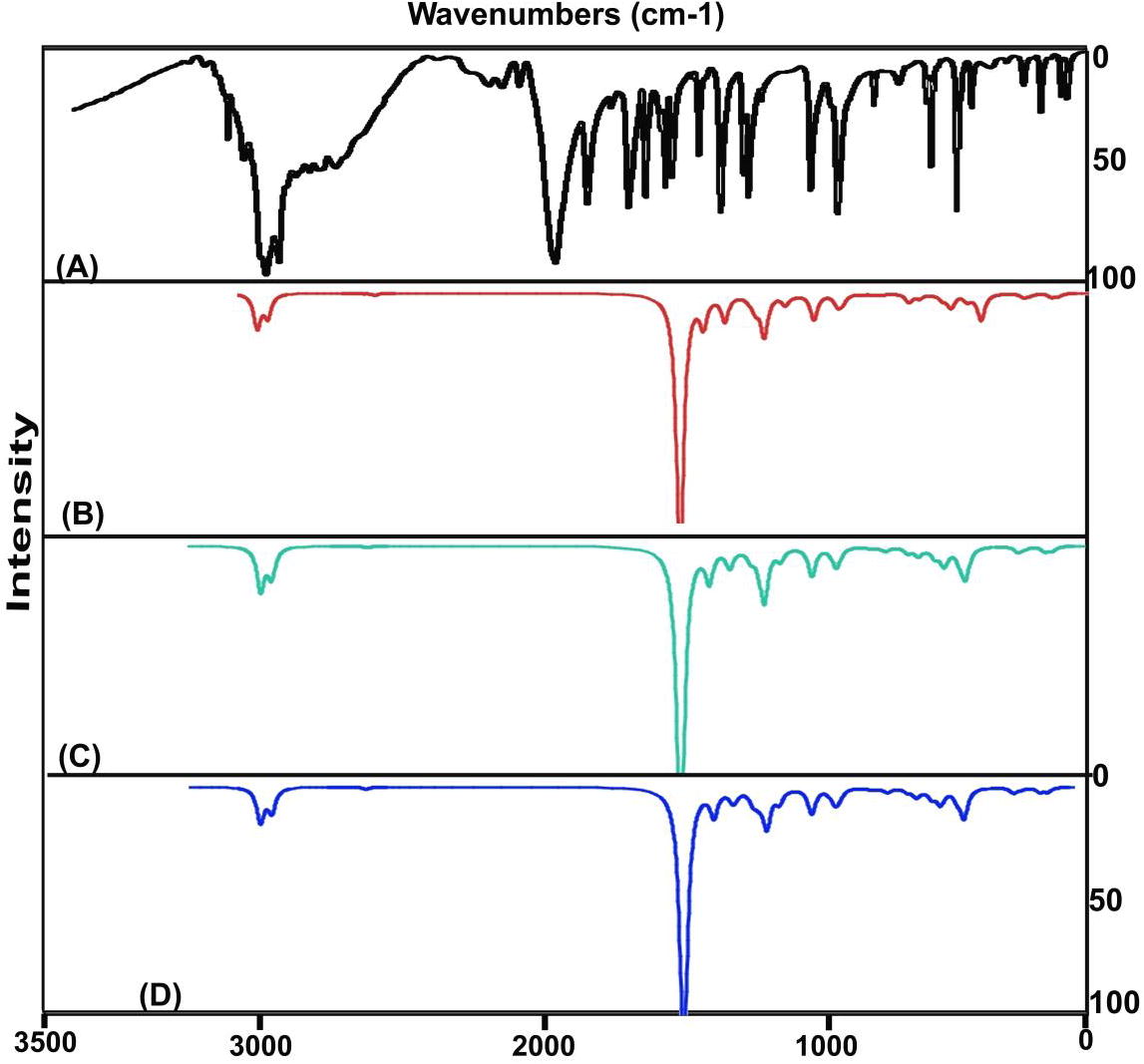
Fig. 5 Experimental and Calculated FT-Raman spectra of Hypoxanthine.
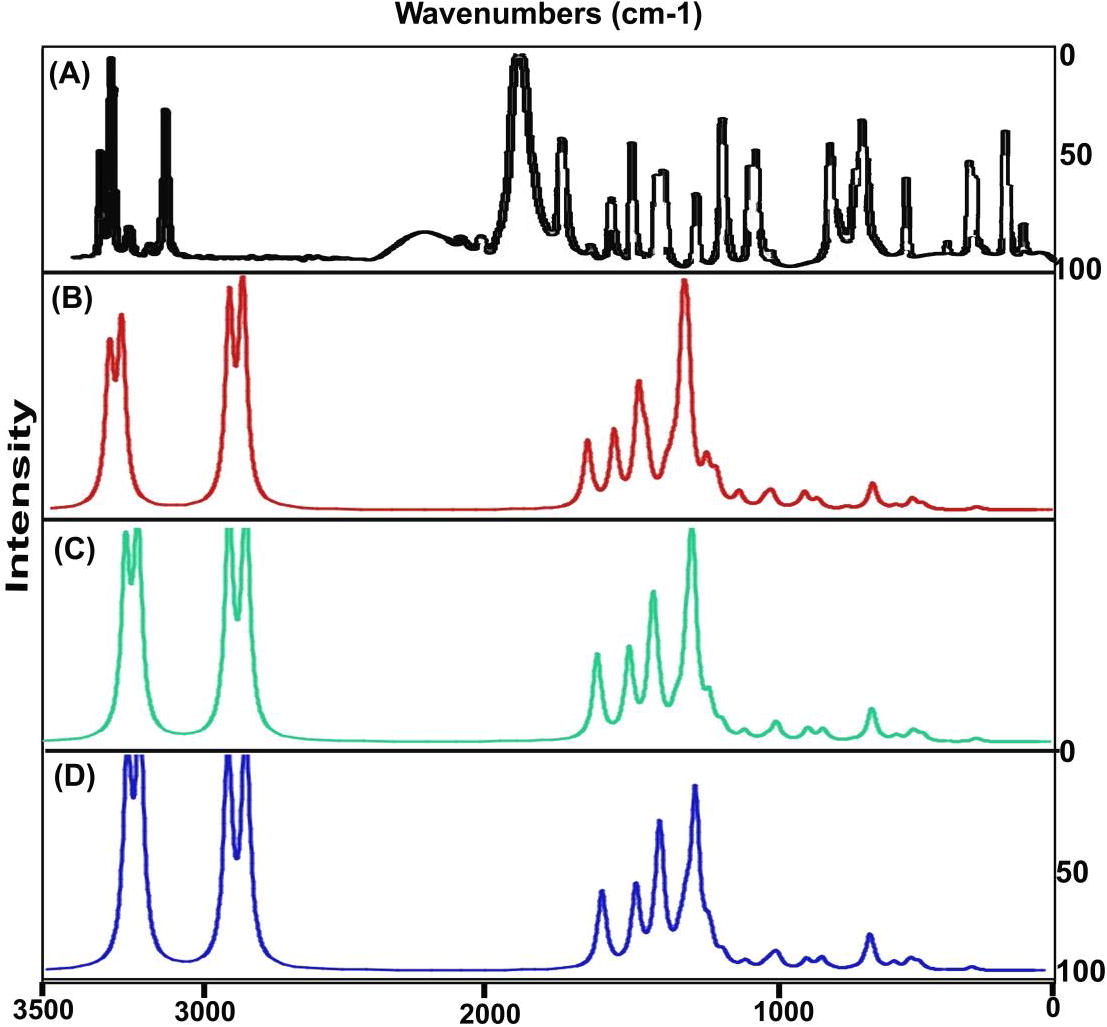
All the vibrational modes were identified and noted with respect to their characteristic region of the spectrum and the entire vibrational modes were found to be obeyed the mutual exclusion principle. The bond length and bond angle vibrations have been assigned in appropriate vibrational region where in which the actual positions of atoms and composed molecular structure were identified.
C-H vibrations
Since the present compound is composed structure of pyridine and Imidazole rings called purine, two C–H bonds were available and to show their corresponding stretching and bending vibrations in appropriate wavenumbers region. As, in general, the purine is not distinguished from pyrimidine [Citation28], the C–H stretching bands are usually appeared in the C–H bands of pyridine compound. These vibrational bands are observed with strong to medium intensity in the region 3120–3010 cm−1[Citation29]. Accordingly, two vibrational bands for C–H stretching were found with strong intensity at 3015 and 3010 cm–1. The related in plane and out of plane bending vibrations were identified at 960 & 890 cm–1 and 650 & 615 cm−1 respectively. These in plane and out of plane bending vibrations are usually observed in the region 1300–1000 cm–1 and 950–700 cm–1 respectively [Citation30,Citation31] . The entire C–H vibrational process were observed to be packed within the characteristics region which explained that, the energy related to these vibrations were consumed obviously and neither not absorbed by other bonds nor spent to other vibrations.
CN and CC vibrations
The ring C=N and C=C stretching vibrational peaks are usually observed in the region 1625–1450 cm−1. Particularly, they observed in the region 1580–1520 cm–1 and 1480–1420 cm–1 respectively for pyridine based derivatives (purine) [Citation32]. Hence, in this case, the C=N vibrational modes were found at 1570 and 1460 cm–1 and C=C stretching band was observed at 1455 cm–1. Here, C=N bands were observed at two dissimilar region since they were positioned in molecule at two different places.
The in plane and out of plane breathing vibrations are normally observed in the region 630–585 cm–1 and 495–375 cm–1 respectively [Citation29]. Here, the fused ring producing ring C–N–C in and out of bending at 580, 570, 550, 530 and 400 cm–1 and 320, 300, 240, 210 and 180 cm–1 respectively. Here, since the pyridine ring adopted with imidazole in the molecule, some of in plane and out of plane bending modes were moved down and crossed the lower limit of the expected region. The fusing effect in compound affected characteristic region of vibrations upon which it was confirmed that, the substitution was injected covalently and strongly coupled with the ring. From this discussion, it was inferred that, the pyrimidine ring is fused with imidazole ring. Consequently, the property also blended with one another and the new drug property; biochemical and pharmacological property was generated.
In the case of aromatic and counterpart; imines =C–N, the strong bands are observed at 1360–1250 cm−1 and 1280–1180 cm−1 [Citation33,Citation34] due to conjugation of the electron pair of the nitrogen in the ring imparting double-bond character to the C-N bond, the aromatic imines absorbing strongly in the first region. Therefore, the C-N stretching signals were found with very strong to medium intensity at 1345, 1340, 1270, 1210, 1200 and 1145 cm–1. All the bands were occupied in wide range of spectrum with strong intensity which showed large amount of chemical potential energy was being used for stabilizing chemical reactivity.
The stretching peak for intermediate bond C–C in the ring was observed at 1130 cm−1 which was identified in lower region when compared to the ring vibrations. This is mainly due to the electron evacuation from the bond.
NH2 group and C=O vibrations
The saturated aliphatic ketones vibration; C=O stretching is usually appeared in the region 1700–1680 cm–1[Citation35] whereas in this case, the stretching mode was found at 1670 cm–1 and its out of plane bending at 100 cm–1. From this appeared signal, the presence of ketonic bond in the particular position was emphasized and its observed range was little bit lowered due to the participation in property production.
The N–H stretching vibrations of imine group derivatives are usually observed at 3400–3300 cm–1[Citation36] and the corresponding in plane and out of plane bending are usually appeared in the region 1590–1500 cm–1 and 750–700 cm–1 respectively. Here the stretching, in plane and out of plane bending bands were found at 3275 &3115 cm–1, 1415 & 1360 cm–1 and 885 & 790 cm–1 respectively. Usually, the domination character of the amine group is revealed and relatively their expected region of vibrational bands are elevated [Citation37]. But, in this case, the entire vibrational bands were suppressed much from the expected region. This is mainly by the electron accumulation in the intra-molecular sites.
NMR examination
The 1H and 13C NMR spectra of aromatic compounds not only used for studying chemical environment of the carbons and also for studying chemical property by measuring chemical shift with respect to the perturbation of magnetic shield of the core atoms. The rate of change of chemical shift of C atoms reflect the shield breaking due to the electron dislocation of neighbouring atoms and directly describe the change of chemical property by the injection of ligand over the linear and cyclic compounds.
The observed chemical shift along with the calculated values in gas and solvent phase were presented in the and simulated spectra were depicted in . In the present molecule, the pyrimidine base was found to be connected with the imidazole ring with the ketonic part. The carbons in both rings were substituted subsequently by nitrogen atoms and one was associated with =O and thus three different chemical environments looked to be there in which chemical shifts have been observed. The observed chemical shift for all carbons in the ring were found in the region 120–154 ppm and the calculated values (116–159 ppm). Both the chemical shifts of the compound were well agreed with literature values [Citation37].
Table 4 Experimental and calculated 1H and 13C NMR chemical shift in Hypoxanthine.
Fig. 6 Experimental and Calculated 13C and 1H NMR spectra of Hypoxanthine.
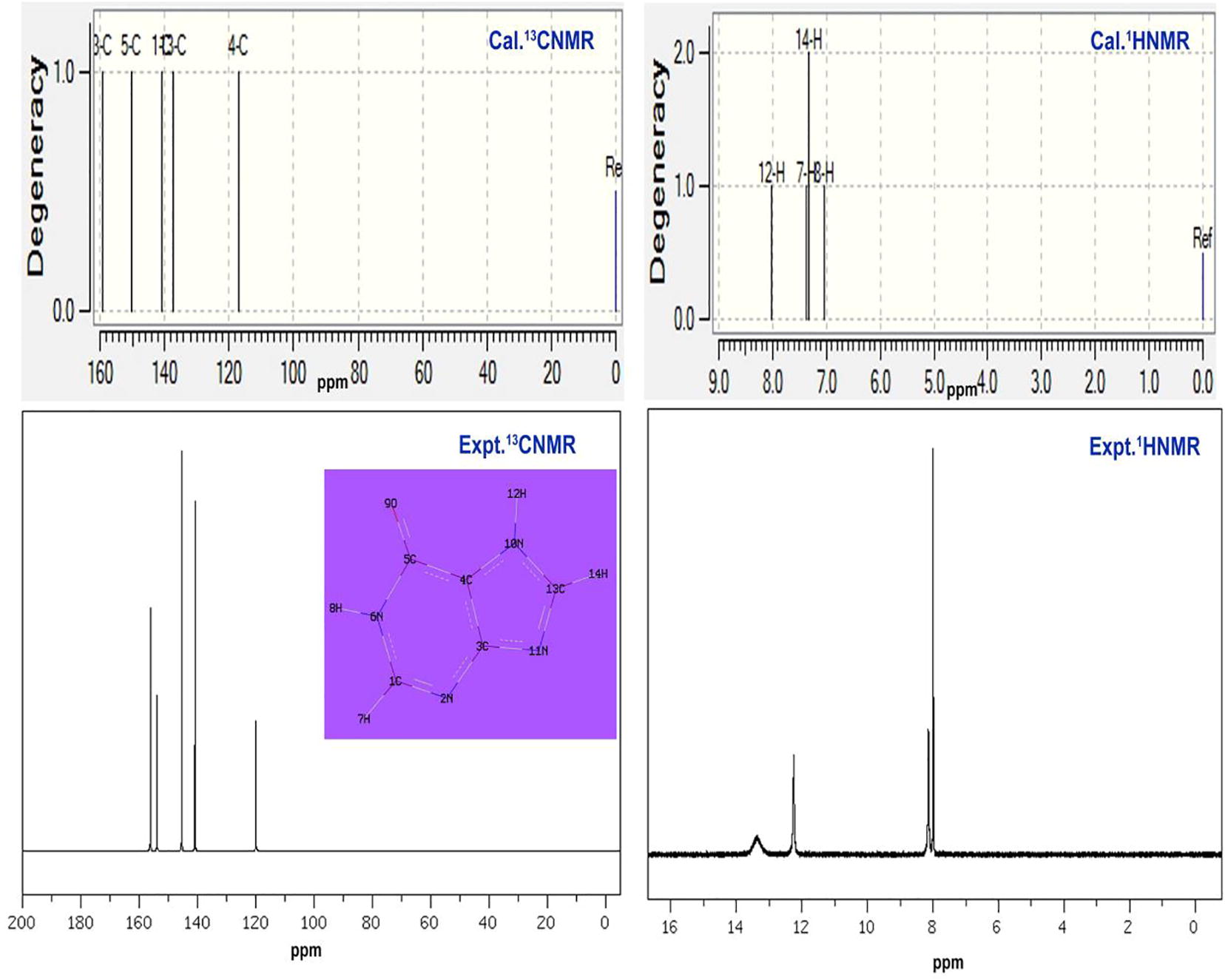
The carbons C1, C3, C4, C5 and C13 in the ring were intercepted by nitrogen atoms and among them, C1 and C13 were having more shift than others due to the abrupt two ways of σ and one way of π -interaction perturbation. Though C1 and C13 were placed on six and five member rings, both the carbons have similar shifts. According to the Mulliken charge analysis, both the carbons having rich positive due to the electron reduction in N and the related bonds called imine group. This circumstances causing the generation of antibiotic property in the compound which is supported by the literature [Citation13]. The chemical shift of ketonic carbon C5 was 145 ppm which is due to the unequal shield breaking by the highly electronegative atom coupling. This carbon was also found to be more negative which showed the important chemical energy transaction in the ring for producing antibiotic action. The chemical shift of C3 and C4 were 141 and 142 ppm and the both the shield breaking was made π- interaction dislocation. From the discussion, it was clear that, the chemical shift was found to be same in two places where the imine groups were placed and the effect of the same was observed. This was the root cause of the inducement of drug property. The other two different observation of chemical shift illustrated additional factor which react more for the stabilization of the drug effect.
Frontier molecular interaction examination
The linear combination of atomic orbitals is usually merged together and structures the frontier molecular orbitals for the aromatic compound and the existence of electrochemical forces among the molecular sites stabilizing orbitals in two sequential ways. They are occupied molecular high energy orbitals (HOMO) and unoccupied low energy orbitals (LUMO). Such arrangement of orbitals are usually influenced by intra molecular interactions where the HOMO and LUMO always being interacted with one another. The energy upon which the construction of overlapping of orbitals between ligand and base compositions are accomplished the resultant physical and chemical property which causing the characteristics and application of the compound. Thus, the transitions between orbitals are complicated and the rate of transitional energy measured the chemical potential to induce desired property. The FMO interaction profile was shown in the and the energy of orbital levels was depicted in the .
Fig. 7 FMO profile of Hypoxanthine.
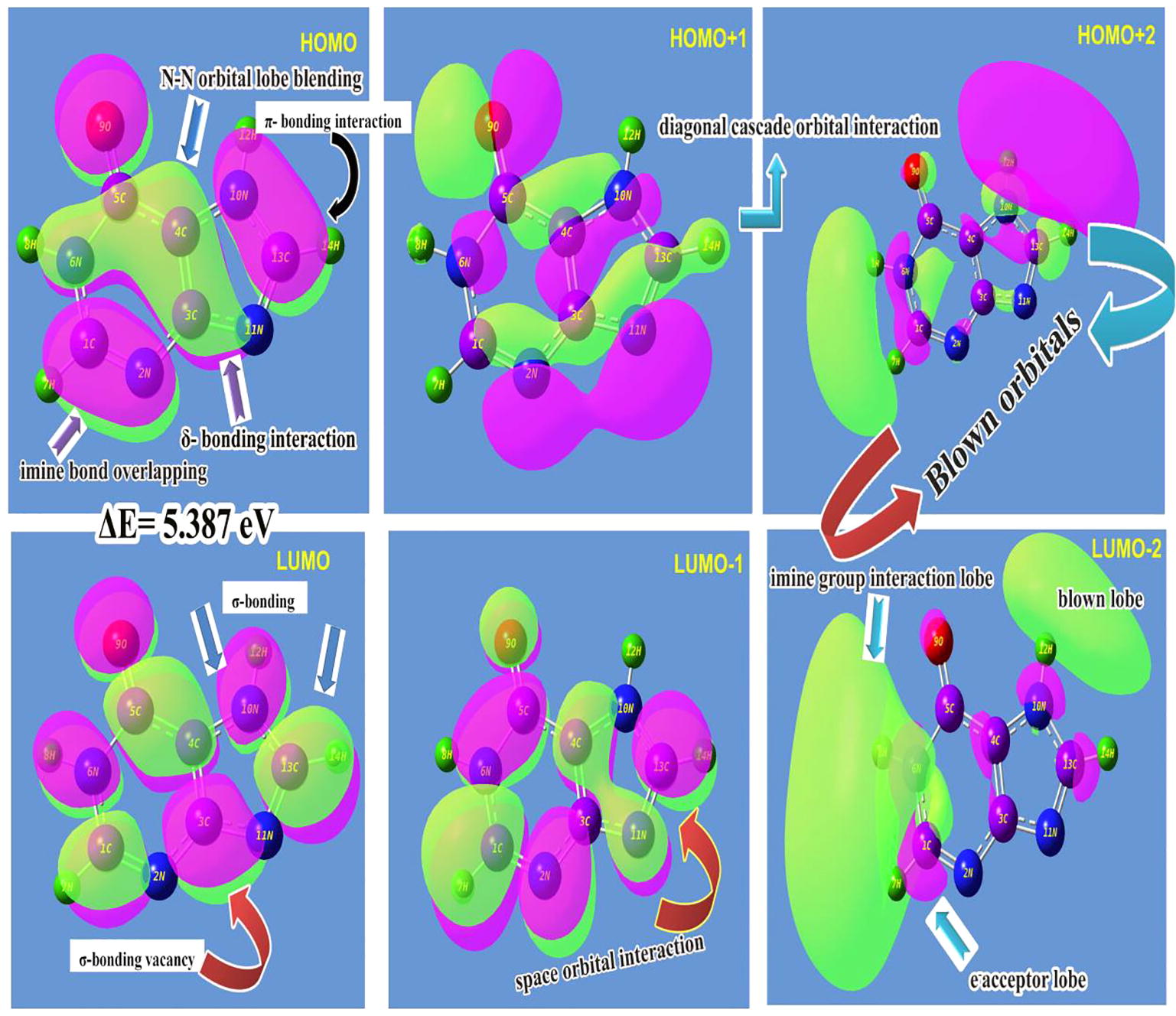
Table 5 Energy levels of Frontier molecular orbital’s of Hypoxanthine.
Here, the HOMO sketch was found to be fully constructed by π-bonding interactions on C=N, C-N and NCCCN. The interacted orbitals were observed over heteronuclear atoms and hyper-conjugation homo-nuclear atoms in the rings which mean that, the electrochemical energy of HOMO are available between five ring bonds for making the transitions in order to induce the drug property. In HOMO+1 level, the cascading of positive and negative interacted iso orbitals were aligned diagonally and are parallel to the plane of the molecular plane. The imine group was played important role in subsequent levels of available energy orbitals also. In HOMO+2 level, the blown orbital interaction was appeared over opposite extreme boundaries of molecule.
In LUMO level, the σ-bonding interactions were occupied and the empty accumulated orbital for receiving the electrochemical potential by the means of electronic transitions from the HOMO level orbitals. The π-bonding interaction was found rarely over the imine group as well as C–H bonds. Here, the empty σ and π- orbitals available for acquiring the electronic energy from energy obtained interaction setup. In LUMO-1 level, the cascading interaction was identified between C4 and N11 where, the degenerate orbitals (space orbitals) were overlapped and the associated potential energy was shared. From these interactions it was concluded that, the available energy domains of HOMO and LUMO were identified and the necessary transitions among them were clearly observed from which the process of sharing chemical potentials were between intra molecular heteronuclear bonds within the ring for inducing drug property were noted.
UV–Visible absorption CT complex profile
The electronic excitation analytical factors of present compound were presented in the and related CT absorption peak diagram of both experimental and simulated were shown in . The electronic absorption peak due to presence of CT complex in the compound is very important in UV–Visible spectra since it provides the information regarding chemical entity which is the root cause of the drug activity [Citation38].
Table 6 Theoretical electronic absorption spectra of Hypoxanthine (absorption wavelength λ (nm), excitation energies E (eV) and oscillator strengths (f)) using TD-DFT/B3LYP/6-311++G(d,p) method.
Fig. 8 UV–Visible spectra of Hypoxanthine.
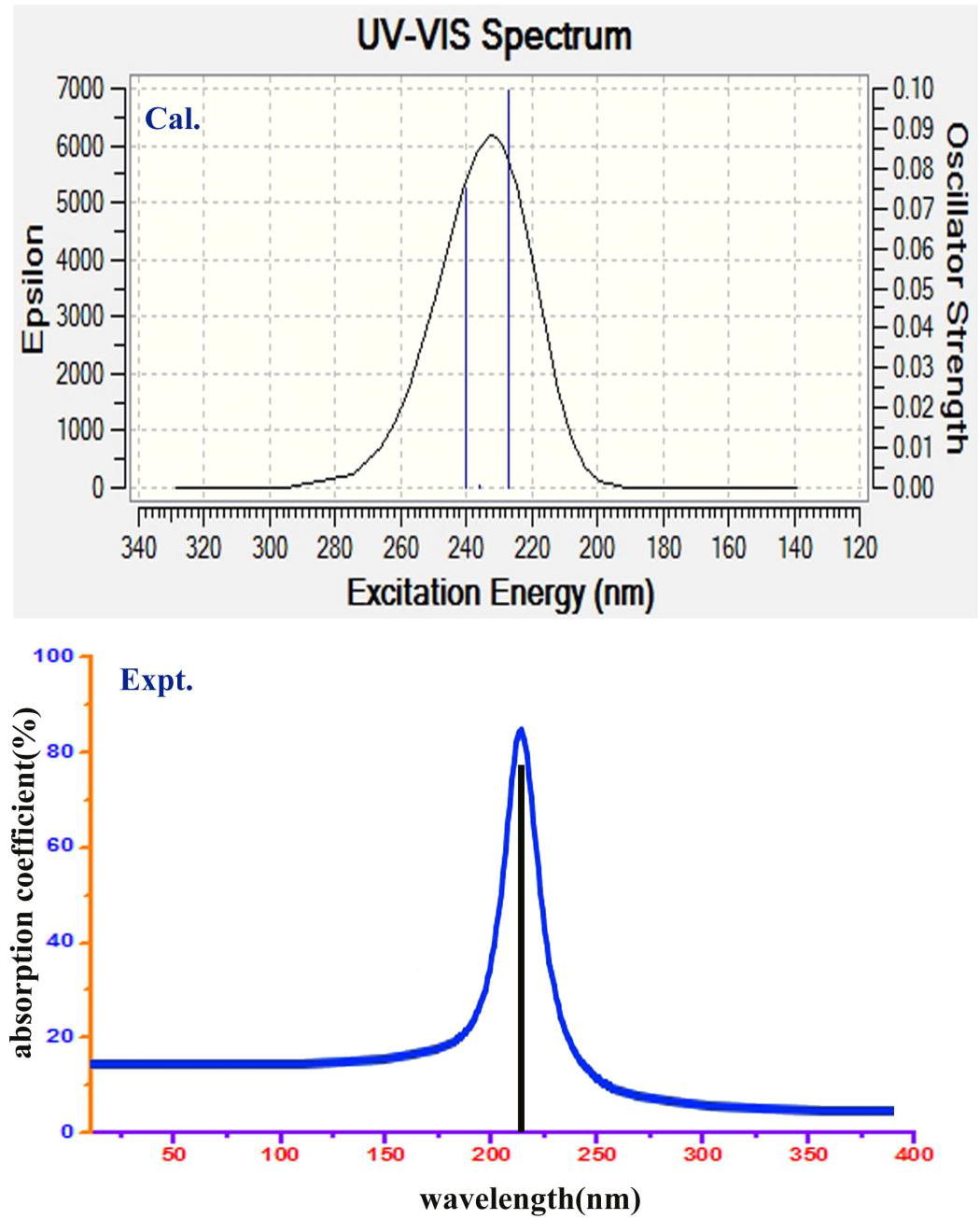
Here, except ketone group, there was no ligand chain found and instead of that, the imidazole ring was fused. According to the and the absorption peak obtained from the electronic spectral region, the CT complex was identified as imine groups which are available in the molecule. The electronic absorption maxima were identified at 240 and 227 nm with 92 and 86% molar efficiency with oscillator strength of 0.07 and 0.09 on the energy gap of 5.15 and 5.45 eV, concurrently, the experimental peaks were observed at 230 with 82% molar efficiency and the entire transitions belong to n → π∗. The observed band was not far from calculated one and the band was (R-band-German, radikalartig) assigned to quartz UV- region of the spectrum. The UV–Visible spectra for present compound were simulated for gas as well as solvent phase (DMSO and CCl4) and there was no difference of impression was obtained by solvent.
As in the , the electronic absorption observed from the transitions gained from the entities associated with imine group. The base ring as well as substitutional ring was holding imine groups over which the charge accumulation was also found. This architecture leads the pharmacological action of hypoxanthine. The presence of ketone group has no effect on property inducement but it controls distortion of drug effect on the molecule and it was emphasized by observing considerable bipolarity. When the CT band is identified in Quartz-UV, the compound will be reactive and biologically active [Citation39]. Accordingly, the chemical reactive mechanism made by CT complex in present compound producing an antibiotic character and it was found consistent.
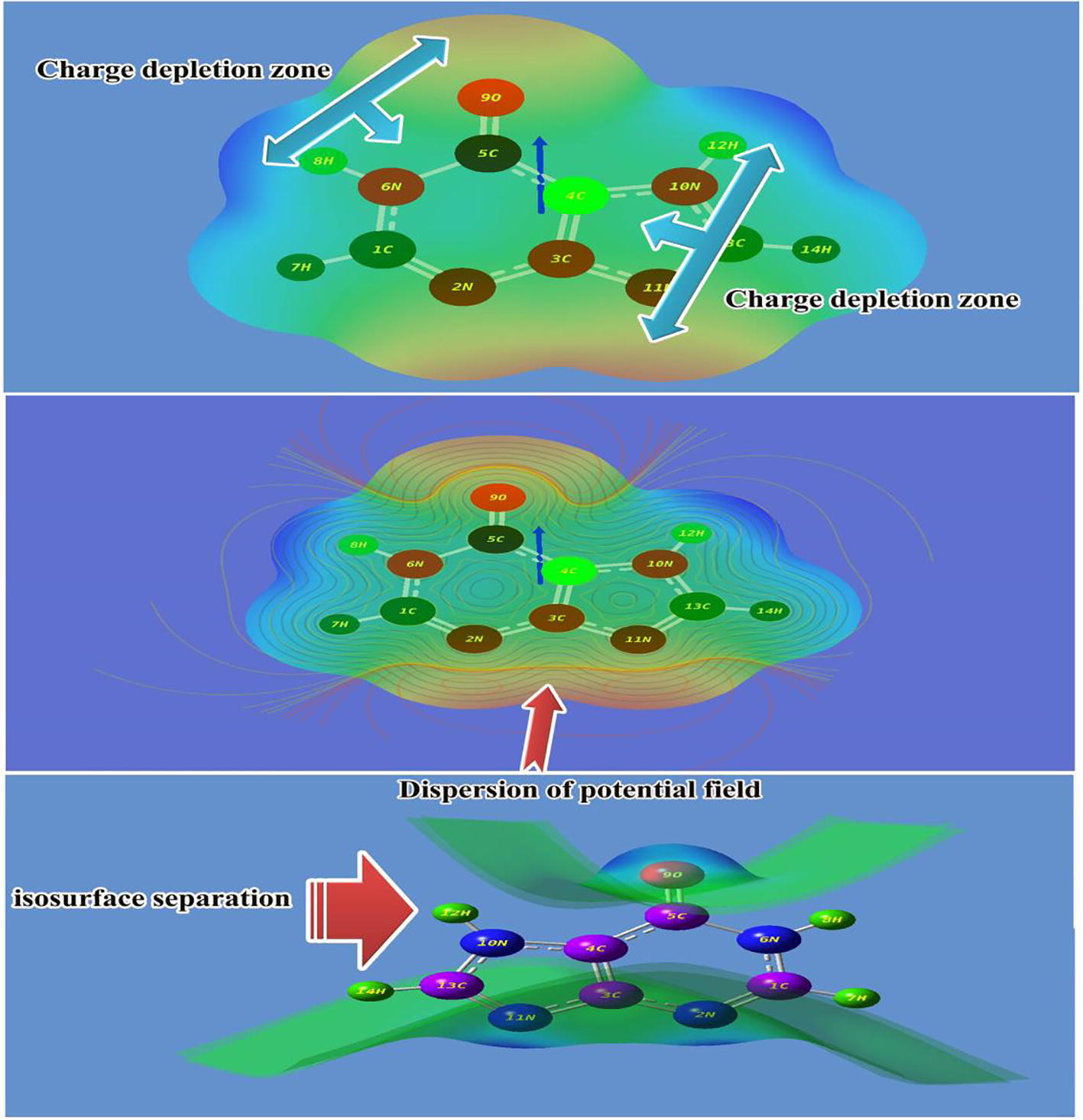
Molecular electrostatic potential (MESP) maps
The Molecular electrostatic potentials (MEPs) usually provided the information regarding chemical reactivity or the biological activity of a compound. The spatial distribution and the values of the electrostatic potential determine the attack of an electrophilic or of a nucleophilic agent as the primary event of a chemical reaction [Citation40]. The three-dimensional nature of the electrostatic potential makes it difficult to simultaneously visualize the spatial distribution and the magnitude of the electrostatic potential. Various approaches have been made to indicate parts of the electrostatic potential. Any point in the space around a molecule is characterized by a specific value of its electrostatic potential. The electrostatic potential on a molecular surface is mainly significant since it is on the molecular surface that molecules come into close contact with chemical reagents or biological receptors [Citation41].
The electrostatic potential map of the present molecule was depicted in between positive and negative potential contours in . Here, for the present molecule, the MESP has been plotted in which the value was identified to be ± 7.843 e−2. In the figure, the positive potential contour was concentrated over the H bonds in the molecule. Consequently, due to the unavailability of the electron clouds, the fields were much reduced in left and right moiety and spreading of potential field was parallel to the plane of the molecule. Wherever the N was present in the compound, the negative potential field contour was concentrated and the enlargement of field lobe was found on top and bottom moiety of molecule. The isosurface separation was determined and the departure field plane was perpendicular to the plane of the molecule. This vision of multi moments of molecule was effective for ligand receptor of proteins. The directions and magnitudes of the dipole moments concentrated over the molecule were supported by charges obtained from a standard Mulliken population analysis [Citation42]. The two charge depletion zones were found between O and C–H bonds of hexagonal structure and N and N–H bond of five membered rings. Such inverse effect of split dipole moments of molecule exposed its good inhibition character.
Fig. 9 MESP view of Hypoxanthine.
Physico-chemical properties
On construction of molecular orbitals, couple of sequence of energized and demotivated or empty orbitals are generated upon which the transitions taking place. From that transitional energy of the orbitals, the physical and chemical properties can be obtained. The calculated parameters were displayed in the .
Table 7 Calculated energies, chemical hardness, electro negativity, Chemical potential, Electrophilicity index of Hypoxathine in UV–Visible region.
The zero point vibrational energy of the present compound in IR and UV–Visible region was found to be −487.13 and −487.14 kcal/mol respectively. The dipole moment of the molecule was calculated by DFT method, found to be 1.66 and 1.68 dyne in IR and UV–visible region respectively. As the dipole moment is the measuring unit of the charge dislocation ability of organic molecule. If it is greater unity, it will be much reactive. Accordingly, it shows the consistent chemical rigidity. The dipole moment was found to be same in UV–Visible and IR region, demonstrated enriched biological activity. The chemical Hardness is one of the highly useful concepts which facilitate chemists to recognize impassivity of the chemical compound. In this case, it was calculated to be 2.72 and 2.83 eV in IR and UV–Visible region respectively. Fundamentally, if the measured value of aromatic compound is greater than unity, the compound will be chemically strong and it will difficult to react. Hence, the observed values showed the rational chemical stability in UV–Visible region.
The force constant of chemical bonds and resultant chemical reactive energy of intra molecular interactions can be measured by the ionization potential [Citation43]. The ionization potential is observed to be 1.37 and 1.18 and if it is greater than unity it will ensure the moderate force constant. Accordingly, the effective values are supposed to be sufficient to maintain the stabilized chemical reactive energy in molecular site. The Electronegativity of a molecule is an important parameter which assesses the local electro-chemical steadiness for the vital application of the prediction of centre of magnetic polarity of molecule [Citation44]. Here, both parameters were found to be 4.09 and 4.02 in IR and UV region respectively. The observed values were found to be enormously high and it is able to bind with protein complex. It also demonstrated the spontaneous accumulation of electron cloud to produce desired drug activity. This effect of the structure also guiding π- bond (C=N) interaction in both the rings and is forcefully verified by MEP diagram.
The electrophilicity index is simply use to estimate of rate of potential energy flow through degenerate interactive molecular orbitals. In this case, the electrophilicity index is 3.07 and 2.85 eV in IR and UV–Visible region respectively. The present compound is the combination of pyridine and imidazole rings and it was noticeably very high which showed the massive quantity of energy approved through overlapped orbitals which is achieved by elevation of electron intensity on C=N groups and was supported by C=O The energy transformation is clearly evidenced from the electrophilicity charge transfer which was found to be + 2.81 for present molecule and it was ensured that, considerable amount of energy was exchanged from pyridine base to imidazole ring and utilized among the bonds to induce the antibiotics activity.
Polarization and hyper polarization analysis
The first order and second order Polarizability display the chemical potential accumulation by means of the arrangement of σ and π- bonding interactions in equilibrium state. When it is irradiated by IR and visible region, it is responded by modulating the irradiated signal by which the hypo and hyper activity of molecular multi moments can be studied. The modulated value can be measured from the Polarizability and hyper Polarizability value of the complex aromatic compound and it is mainly used to predict the molecular properties and biological activities [Citation45]. The higher order methods of computed Polarizability and first order hyperpolarizability indices were depicted in .
Table 8 The dipole moments µ (D), the polarizability α(a.u.), the average polarizability αo (esu), the anisotropy of the polarizability Δα (esu), and the first hyperpolarizability β(esu) of Hypoxanthine.
The calculated average Polarizability and anisotropy of the Polarizability of the present compound was 109 × 10−30esu and 130 × 10−30esu respectively and the hyperpolarizability (β) was found to be 95.32 × 10−33esu. Usually the first and second order Polarizability of the aromatic compound are differenced much since, the hyper action of the molecular multi moments are always higher than hypo activity of the molecular moments [Citation46]. But, here, both the values were found to be close to each other and it was due to the first order produced the antibiotic character and it was stabilized by second order Polarizability. It is very rarely observed in aromatic compounds by which the novel drug property substantiated [Citation47]. This was achieved by the fusing of two rings and most of enriched antibiotic compounds would be the derivatives of this structure. From this discussion, it was concluded that, since the compound having hyper symmetrical polarizations, it was strongly approved the pharmaceutical property.
NBMO transition analysis
The chemical potential energy that is consumed by different identities of the molecule for fabricating peculiar drug property can be measured by the individual transitions among polarized non bonding orbitals. The concise quantity of electronic energy is operated the transitions (charge-transfer) between donor and acceptors of various entities to setup the desired chemical mechanism related to drug property [Citation48]. Accordingly, in which places, the consistent amount of energy used for the attainment of drug property can be studied from energy transitions in Lewis structure and were tabulated in .
Table 9 The calculated NBO of Hypoxanthine by second order Perturbation theory.
Here, the several transitions have been observed over entire compound among which some of the transitions causing major drug property have been identified. In the first case, within the six membered ring, the energy of 5.18 and 6.27 kcal/mol were transferred from C1-N2 to C3 and C3-N11 respectively which were assigned as π – π∗ interaction system. Similarly, the energy of 3.25 and 2.95 kcal/mol was found to be transferred from C3-C4 to N2 and N2-C3 and was assigned as π – π∗. Another transitions have been observed from C3-C4 to N2-C3 and C4-N10 in which 5.22 and 3.09 kcal/mol respective energy were used for σ-σ∗ and π – π∗ respectively. The important transitions from C4-C5 to C3-C4 and C4-N10 were observed for consuming 6.74 and 4.43 kcal/mol energy between two rings.
The transitions C4-N10 to N2-C3 and C3-C4 were observed in reverse order for taking energy 4.76 and 3.15 kcal/mol. Similarly, the transitions C5-N6 to C4-N10 by consuming 4.53 kcal/mol amount of energy and was assigned as σ-σ∗. The second order transitions from N10–C13 to C4-C5 and from N11-C13 to N2-C3 were observed by taking 6.77 and 9.78 kcal/mol which was assigned to σ-σ∗. The important transitions from lone pair of N2 to C1, C4 to C3, N6 to C1, N2 to C3, N6 to C1 and O9 to C5 by absorbing energy of 3.1, 3.4, 5.9, 3.1, 3.9 and 19.10 kcal/mol respectively. Here, the large number of lone pairs has participated in which the considerable amount of energy was used for the energy transitions. Here, the important transition has been observed in NMBO of ketonic group whereas the there was no important effect was observed in bonding process. Here, chemical mechanical path was identified from C3 to C4 via N11, C13 and N10 similarly, another root was observed from C3 to C4 via N2, C1, N6 and C5. Here, the C3-C4 was acted as central terminal for chemical process according which, the path way for inducement of chemical mechanism for generating the drug property was identified. From this observation, it was clear that, fusing of rings themselves behaved as good drug.
Thermodynamic analysis
Since thermodynamic parameters; entropy, specific heat capacity and enthalpy were found to be varying absolutely, the endothermic reaction has positive temperature coefficient whereas the Gibbs free energy was identified to be varied negatively because of the continuity was inverse. Due to this predictable characteristics emphasize the compound to be chemically strong, the unique chemical property and having endless chemical reaction.
The entropy is a measure of dissociation of chemical system under the perturbation of internal energy with respect to temperature. It happened to be observed elevate mode which showed the enriched chemical process. Similarly, the enthalpy is the measurement of the heat content of molecular system absorbed from external [Citation49]. Usually, the ΔH is a positive change in endothermic reactions according which, in this present case, the chemical reaction was found to be conserved and the considerable potential energy consumed for fusing both the rings. The specific heat capacity of the present compound also found to be increased with respect to temperature. The kinetic energy of the system of molecules was ultimately developed and strong chemical system was formed and it was clear that, the process outsopened kinetics of internal parts to be active.
VCD profile
The simulated VCD spectrum for optimized structure was displayed in the . Each and every transitional structure having specific VCD spectrum and the conformational structure has unique spectrum which represent the chiral character of the molecule under study. The IR and Raman spectral sequence also supported by VCD spectrum and used to study the chirality character and thereby comfortable to explain whether the compound is biologically and pharmaceutically active with minimized toxicity or not. Usually, the symmetrical sequence of spectrum in absorption as well as transmission reflected the less toxicity effect of the compound [Citation50]. Accordingly, from the spectrum, it was observed that, the present compound having progression VCD pattern which leads to contain masked side effect. Here, the observed peaks with sequential pattern on both side (absorption and transmission)(±0.06) identified in the VCD spectrum showed that, the compound itself will be acted as good drug with enlarged chemical purity (see ).
Fig. 10 VCD spectrum of Hypoxanthine.
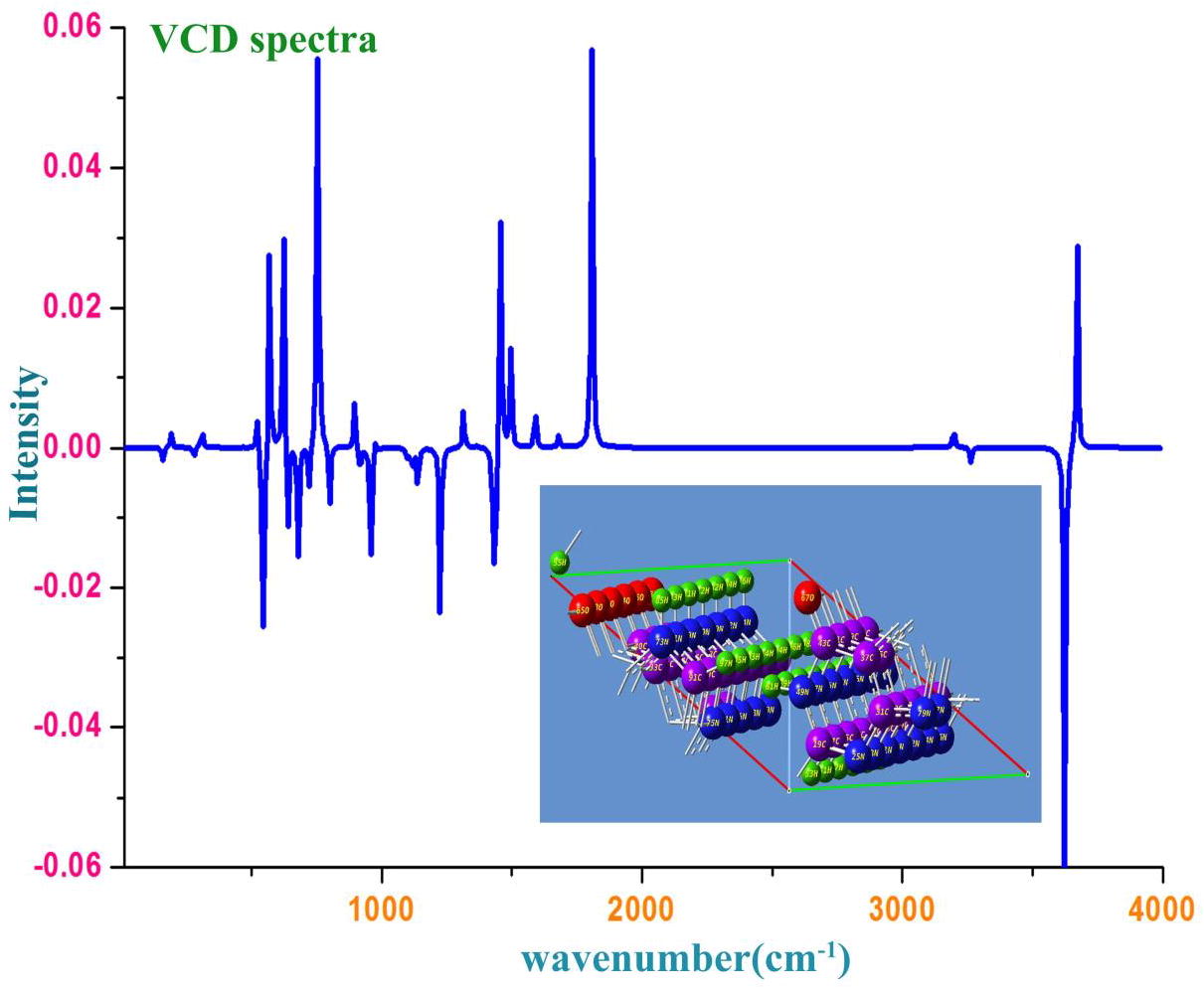
Table 10 Specific heat capacity, entropy and enthalpy at different temperatures for Hypoxanthine.
Conclusion
The present compound was the composition of pyrimidine and imidazole rings and basically its derivatives was found to be good drug. Moreover, since, the present compound was found to be drug itself, the rate of chemical property for the cause of drug activity was investigated. Accordingly, in this work, several analyses have been made which symbolized the drug activity and the entire experimental and theoretical studies proved that, the present compound was exposed its drug property and was ensured from the literature work. Here, the compound has many imine functional groups which are the main source of drug behaviour. It was also ensured by frontier molecular energy transition, electronic CT complex analysis and NMBO profile. The biological activity parameter analysis has also proved goodness of the drug. The compound itself without substitutions was acted as antibiotic drug and if the suitable substitutions added in appropriate places, the molecular complex will may be acted as good anticancer drug.
Acknowledgement
The Authors thank the computational consultancy, A.V.C. College (Autonomous), Mayiladuthurai, Tamilnadu, India and co-computational consultancy service headed by Dr. K. Senthil Kannan, Dean R & D, EDAYATHANGUDY G.S. PILLAY ARTS & SCIENCE COLLEGE, Nagapattinam, Tamilnadu, India.
References
- M.Fernandez-QuejoM.de la FuenteM.NavarroTheoretical calculations and vibrational study of hypoxanthine in aqueous solutionJ Mol Struct7442005749757
- JRLustyCRC Handbook of Nucleobase ComplexesVol. I1990CRC PressBoca Raton, FL
- J.S.KwiatkowskiT.J.ZielinskiR.ReinP.O.LowdinAdvances in quantum chemistry181986Academic PressNew York8592
- K.S.JainT.S.ChitreP.B.MiniyarM.K.KathiravanV.S.BendreV.S.VeerS.R.ShahaneC.J.ShishooBiological and medicinal significance of pyrimidinesInt J Org Bioorg Chem41201415
- F.L.RodneyG.CharlesSkinnerS.WilliamFused pyrimidines. The heterocycle of diverse biological and pharmacological significanceCan J Chem45196722132216
- V.P.LitvinovAdvances in heterocyclic chemistry922006Russian Academy of Sciences119991 Moscow, Russia 83
- Samar SaidFatahalaSherifaHasabelnabyAymanGoudahGhada.MahmoudRaniaHelmy Abd-El HameedPyrrole and fused pyrrole compounds with bioactivity against inflammatory mediatorsJ Mol224612017118
- R.RamaekersG.MaesL.AdamowiczA.DkhissiHybrid density functionals and ab initio studies of 2-pyridone–H2O and 2-pyridone–(H2O)J Mol Struct5602001205221
- W.SaengerAdvanced text in chemistry, principles of nucleic acid structure1984SpringerNew York p. 164
- B.HernandezF.J.LuqueM.OrozcoAdvances in heterocyclic ChemistryJ Org Chem6119965964
- R.M.IzattJ.J.ChristensenJ.H.RyttingSites and thermodynamic quantities associated with proton and metal ion interaction with ribonucleic acid, deoxyribonucleic acid, and their constituent bases, nucleosides, and nucleotidesChem Rev7151971439481
- E.CasassasR.GargalloI.GiménezA.Izquierdo-RidorsaR.TaulerStudy of the acid-base behavior and copper (II) complexing properties of uracil- and hypoxanthine-derived nucleotides in aqueous solutionJ Inorg Biochem5631994187199
- N.MoorthyP.C.JobePrabakarS.RamalingamM.GovindarajanS.Joshua GnanamuthuG.V.PandianSpectroscopic analysis, AIM, NLO and VCD investigations of acetaldehyde thiosemicarbazone using quantum mechanical simulationsJ Phys Chem Solids9520167488
- S.XavierS.PeriandySpectroscopic (FT-IR, FT-Raman, UV and NMR) investigation on 1-phenyl-2-nitropropene by quantum computational calculationsSpectrochim Acta Part A Mol Biomol Spectrosc1492015216230
- A.MadanagopalS.PeriandyP.GayathriS.RamalingamS.XavierVibrational and NMR investigation on pharmaceutical activity of 2,5-dimethoxy-4-ethylamphetamine by theoretical and experimental supportJ Mol Pharm Org Process Res512017122
- J.S.SinghFT-IR and Raman spectra, ab initio and density functional computations of the vibrational spectra, molecular geometries and atomic charges of uracil and 5-halogenated uracilsSpectrochim Acta A Mol Biomol Spectrosc117C2013502518
- M.S.IslamA.M.Al-MajidA.BarakatS.M.SolimanSynthesis, molecular structure and spectroscopic investigations of novel fluorinated spiro heterocyclesMolecules205201582238241
- V.BalachandranK.ParimalaMolecular structures, FT-IR and FT-Raman spectra, NBO analysis, NLO properties, reactive sites and quantum chemical calculations of keto-enol tautomerism(2-amino-4-pyrimidinol and 2-amino-pyrimidine-4(1H)-one)Spectrochim Acta A Mol Biomol Spectrosc10220133051
- Cong-MingZhangXi-FengGaoMeiZhuYun-GaiLiQing-LunWangLi-CunLiSynthesis, crystal structure and magnetic characterization of two Mn(III) chains with Schiff-base ligandsJ Mol Struct10332013813
- Hossein PashaEbrahimiJabbar S.HadiTahseen A.AlsalimThaer S.GhaliZeinabBolandnazarA novel series of thiosemicarbazone drugs: from synthesis to structureSpectrochim Acta Part A Mol Biomol Spectrosc137201510671077
- M.MellaouiS.BelaidiD.BouzidiN.GuerrafStructure activity relationship and quantitative structure-activity relationships modeling of antitrypanosomal activities of alkyldiamine cryptolepine derivativesJ Comput Theor Nanosci12920152127
- A.KerassaS.BelaidiT.LanezComputational study of structure-property relationships for 1,2,4-oxadiazole-5-amine derivativesQuant Math520164552
- Z.Imane AlmiS.BelaidiL.SegueniComputational exploration in the electronic structure property/activity relationships and multi-parameter optimization of 1,2,5-thiadiazole derivatesJ Rev Theor Sci32015310
- D.F.VeberS.R.JohnsonH.Y.ChengB.R.SmithK.W.WardK.D.KoppleMolecular properties that influence the oral bioavailability of drug candidatesJ Med Chem4512200226152623
- P.ErtlB.RohdeP.SelzerFast calculation of molecular polar surface area as a sum of fragment-based contributions and its application to the prediction of drug transport propertiesJ Med Chem43200037143717
- C.A.LipinskiF.LombardoB.W.DominyP.J.FeeneyExperimental and computational approaches to estimate solubility and permeability in drug discovery and development settingsAdv Drug Deliv Rev461–32001326
- J.H.ListerChemistry of heterocyclic compounds fused pyrimidines. Part II: PurinesJ Heterocycl Chem71971496502
- GeorgeSocratesInfrared and Raman Characteristic Group Frequencies (Tables and Charts)2001John Wiley & Sons Ltd60124
- Y.XueD.XuD.XieG.YanSpectroscopic properties of inorganic and organometallic compoundsSpectrochim Acta A Mol Biomol Spectrosc5620001929
- Y.XueD.XieG.YanHybridization: a chemical bonding nature of atomsInt J Quant Chem762000686689
- Ardyukova AF, Shkurko OP, Sedova VF. Atlas of spectra of aromatic and hetrocyclic compounds. Infrared spectra of pyrimidine series, vol. 4. Novosibirsk: Nauka Sib. Otd.; 1974. p. 1–128.
- Pouchert CJ, The Aldrich Library of FTIR Spectra, vol. 1:1, The Aldrich Co., 1985. p. 1–254.
- K.OhnoK.MorokumaF.HirotaH.HosoyaS.IwataY.OsamuraH.KashiwagiS.YamamotoInfrared and Raman characteristic group frequencies: tables and chartsJ Mol Struct26819924150
- P.J.KrugerSpectroscopic studies of hydrogen bondingCan J Chern5119731363
- Scholarly articles for J Fabian Bull Soc Chim Fr Semi empirical calculations on sulfur-containing heterocycles. J Phys Chem 1968;72(12):3975–85.
- S.RamalingamS.PeriandyS.MohanVibrational spectroscopy (FTIR and FTRaman) analysis on 2-Bromo-4-methylaniline using HF and DFT calculationsSpectrochim Acta A Mol Biomol Spectrosc77120107381
- N.MoorthyP.C.JobePrabakarS.RamalingamG.V.PandianP.AnbusrinivasanVibrational, NMR and UV-visible spectroscopic investigation and NLO studies on benzaldehyde thiosemicarbazone using computational calculationsJ Phys Chem Solids9120165568
- Robert S.MullikenMolecular compounds and their spectra. IIACS PublicationJ Am Chem Soc741952811824
- M.Manzoor AliGeneGeorgeS.RamalingamS.PeriandyV.GokulakrishnanVibrational [FT-IR, FT-Raman] analysis, NMR and mass – spectroscopic investigation on 3,6-dimethylphenanthrene using computational calculationJ Mol Struct10992015463481
- John D.WestbrookRonald M.LevyKarstenKrog-jespersonMolecular electrostatic potentials and partial atomic charges from correlated wave functions: applications to the electronic ground and excited states of 3-methylindoleJ Comput Chem13131992979989
- R.S.BohacekC.McManinRepresentation of molecular electrostatic potentials by topologicalJ Mid Chem35199216711684
- R.S.MullikenElectronic population analysis on LCAO–MO molecular wave functionsJ Chem Phys23195518331838
- Moore CE, Ionization potentials and ionization limits derived from the analysis of optical spectra. NSRDS-NBS 34. Washington, DC, 1970.
- Tro, Nivaldo J. Chemistry: a molecular approach, 2nd ed., New Jersey; 2008.
- J.WangX.Q.XieT.HouX.XuFast approaches for molecular polarizability calculationsJ Phys Chem A11120200744434448
- N.MoorthyP.C.Jobe PrabakarS.RamalingamS.PeriandySpectroscopic investigation of the stimulus of NLO property on acetone thiosemicarbazone using computational [HF and DFT] confinementJ Theor Comput Sci220154
- CorwinHanschWayne E.SteinmetzAlbert J.LeoSuresh B.MekapatiAlkaKurupDavidHoekmanOn the role of polarizability in chemical–biological interactionsJ Chem Inf Comput Sci432003120125
- Gao-fengYuanBoSunJingYuanQiao-meiWangEffects of different cooking methods on health-promoting compounds of broccoliJ Zhejiang Univ Sci B1082009580588
- Brij Lal, Subramanian N. Heat and thermodynamics. 4th ed.. S Chand Publications; 2006. p. 197.
- P.J.StephensF.J.DevlinJ.R.CheesemanCircular dichroism and stopped-flow spectrometers. Chirascan Systems, Stopped-Flow Systems, VCD spectroscopy for organic chemists2012CRC Press Taylor & Francis Group 25