
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
In this paper a complete quantum chemical study of [1,2,4]-triazolo [3,4-b][1,3,4] thiadiazole has been done with the combination of DFT/B3LYP method and 6-311G(d, p) basis set. The vibrational assignments are calculated with the help of PED. By using quantum theory of atoms in the molecule (QTAIM) we have calculated topological parameters at BCP point by which the nature of several intermolecular hydrogen bondings are studied. Electronic properties are calculated with the help of HOMO-LUMO plot. Chemical active sites of title molecule are described by FF, chemical hardness, chemical softness etc. UV spectra are calculated with the help of TDDFT using optimized parameters. NBO analysis gives idea about transfer of charge between bonding and antibonding electrons. Biological activity analysis suggests that the molecule can be used in anti- inflammatory drugs to explore new drugs. The molecule is docked with MBNL1 receptor with the help of Swiss-Dock online server with Full fitness score of −541.58 a.u.
1 Introduction
1,3-thiazole containing both Nitrogen and sulphur is a heterocyclic compound. The term 'thiazole' refers to a large family of derivatives [Citation1]. Thiazole having molecular formula C3H3NS is a soft yellow liquid with pyridine-like scent [Citation2] and used in a variety of specialized products, often by fusing with benzene derivatives. These are naturally occurring peptides, and utilized in the commercial development of peptidomimetics [Citation3,Citation4] . Thiazole is heterocyclic series organic compound characterized by a ring structure composed of three carbon atoms, one nitrogen atom, and one sulphur atom. This ring structure attributes many significant biologically active natural qualities such as thiamine, penicillin, and in many synthetic drugs, dyes, and industrial chemicals [Citation5,Citation6] . These are found in a number of various food products and flavors and are used in the manufacturing of prepackaged food products including noodles [Citation7]. Thiazoles help flavorists to create healthier crops by enhancing the flavor of meats and savory foods without the accumulation of excess salts or fats [Citation8]. 1,2,4-Triazole derivatives and their bonded heterocyclic correspondents are well known for their different biological activities and 1,2,4-triazole rings have been combined into ligands used in coordination compounds. The 1,2,4-triazole derivatives and their N bridged heterocyclic equivalents have been widely studied [Citation9Citation[10]Citation[11]Citation[12]Citation[13]–Citation14] . Numerous 3,6-disubstituted [1,2,4] triazole [3,4-b] [1,3,4] thiadiazoles are testified to display significant antibacterial [Citation15Citation[16]Citation[17]–Citation18] , pesticidal [Citation19], anticancerous [Citation20], anti-inflammatory, and anti-oxidant activities [Citation21]. Many studies are carried to synthesize organic compounds, inorganic metal complexes and are detected by using NMR FTIR spectra [Citation22Citation[23]Citation[24]Citation[25]Citation[26]–Citation27] . Monirah A. Al-Alshaikh etal. synthesized the crystal structure of [1,2,4]-triazolo[3,4-b][1,3,4] thiadiazole molecule and found it as a potential bioactive agent [Citation22Citation[23]Citation[24]Citation[25]Citation[26]Citation[27]–Citation28] . The present study on the [1,2,4]-triazolo[3,4-b][1,3,4] thiadiazole molecule has been carried out to support and as an extension of the work of Monirah A. Al-Alshaikh et al. [Citation29]. As mentioned in the work of Monirah A. Al-Alshaikh et al., due to bioactive potential of the molecule its biological properties are studied by using several parameters and also docking is made for designing new anti-inflammatory drugs. Along with this the geometrical parameters, NMA, Biological and Chemical activities, electronic transitions, thermodynamic properties are studied by the combination of DFT/B3LYP method and 6-311G(d, p). A complete DFT analysis is performed on the title molecule ().
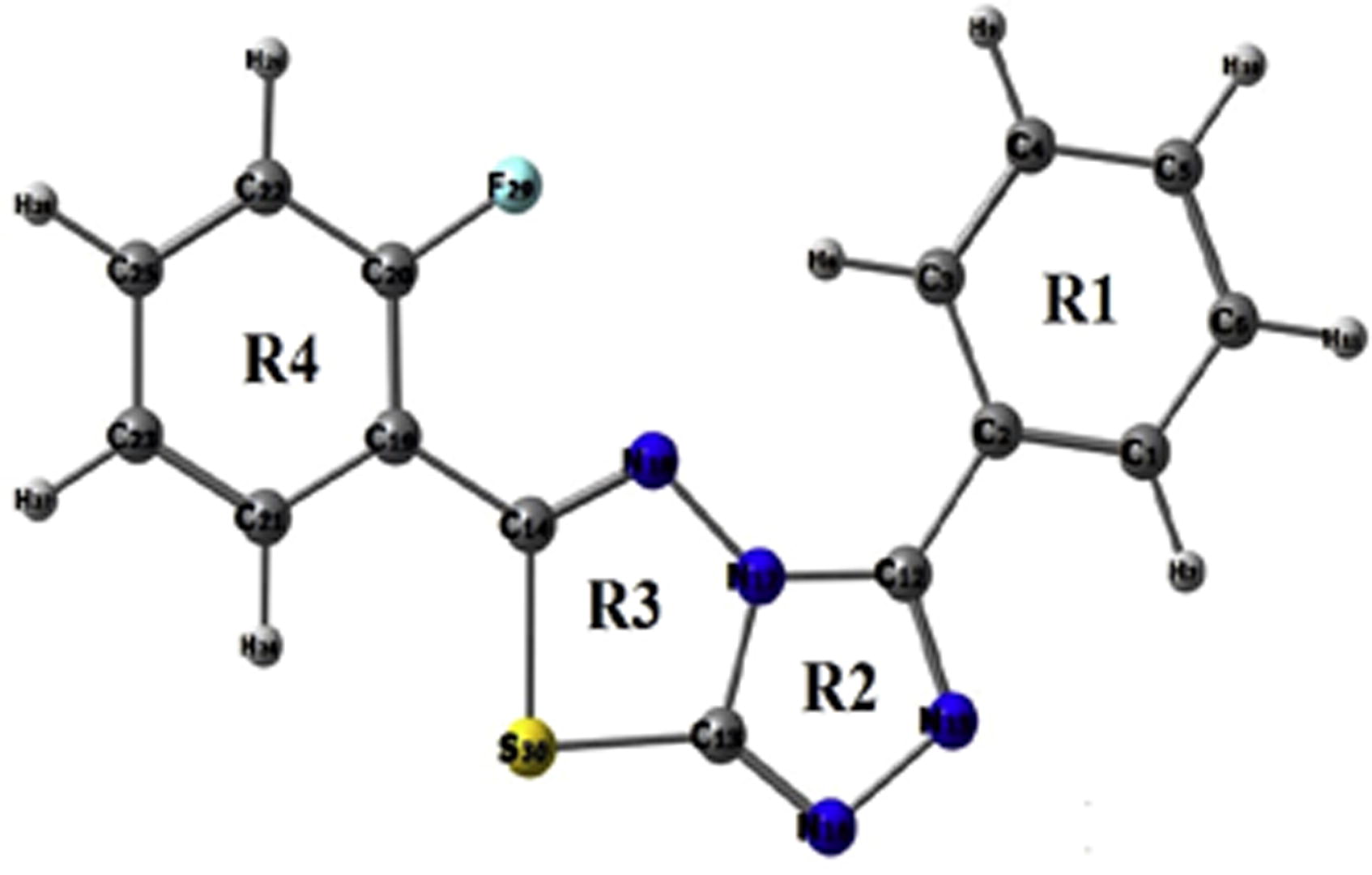
Fig 1 Molecular Picture of title molecule according to numbering.
2 Computational details
All the calculations are done with G03 program package [Citation29] using combination of B3LYP/DFT method and 6-311G (d, p) basis set. This basis set 6-311G (d, p), with ‘p’ polarization functions on hydrogen atoms and ‘d’ polarization functions on heavy atoms, is used for better description of polar bonds of molecule [Citation30,Citation31] . Initial geometry is modeled with the help of Gauss View 3.0 program package [Citation32]. AIM calculation, performed by AIMALL program package [Citation33], is based on quantum theory of atoms in a molecule. Natural bond analysis is carried with NBO 3.0 program package [Citation34]. Electronic transitions, vertical excitation energies, and oscillator strengths are computed with the TD-DFT method.
3 Result and discussion
3.1 Optimized parameters
The molecule taken has two benzene rings, one thiazole and other phenyl rings. The molecule shows some deviation from planarity, with fluoro ring (C19-C14) forming dihedral angle of 2.65° and the phenyl ring (C12-N17) 3.84°. The distance of the interaction between benzene ring and thiazole ring C14-C19 is 2.33 Å.
AIM [Citation35] calculation shows that there are two interactions between N18-H8 and N18-F29 in the molecule, which are shown in . According to Koch and Popelier standard, an existence of hydrogen bond follows bond critical point (BCP) for the ‘proton (H)…acceptor (A)’, [Citation36] which involves electron density (q) in the range of 0.002–0.040 a.u. and Laplacian () in the of range 0.024–0.139 a.u. All calculated parameters at BCP, in between N18-H8, and N18-F29, are listed in . On the basis of topological parameters three types of H-bonds are characterized. According to Rendering to Rozas et al. [Citation37], the characterization demands, at BCP, that for strong and covalent nature of H-bond
< 0 and H < 0, for medium and partially covalent nature of H-bond
> 0 and H < 0, and for weak H-bond of electrostatic character
> 0 and H > 0. From , the Laplacian of charge density are positive, however value of H < 0 signifies that the interaction is medium strong in nature. According to Espinosa et al. interaction energy calculated is Eint = ½ V at BCP [Citation38]. In this study calculated interaction energy of N18-H8, N18-F9 is 3.95 kcal/mol and 5.648 kcal/mol respectively.
Fig 2 Molecular Graph of the studied compound at BCP.
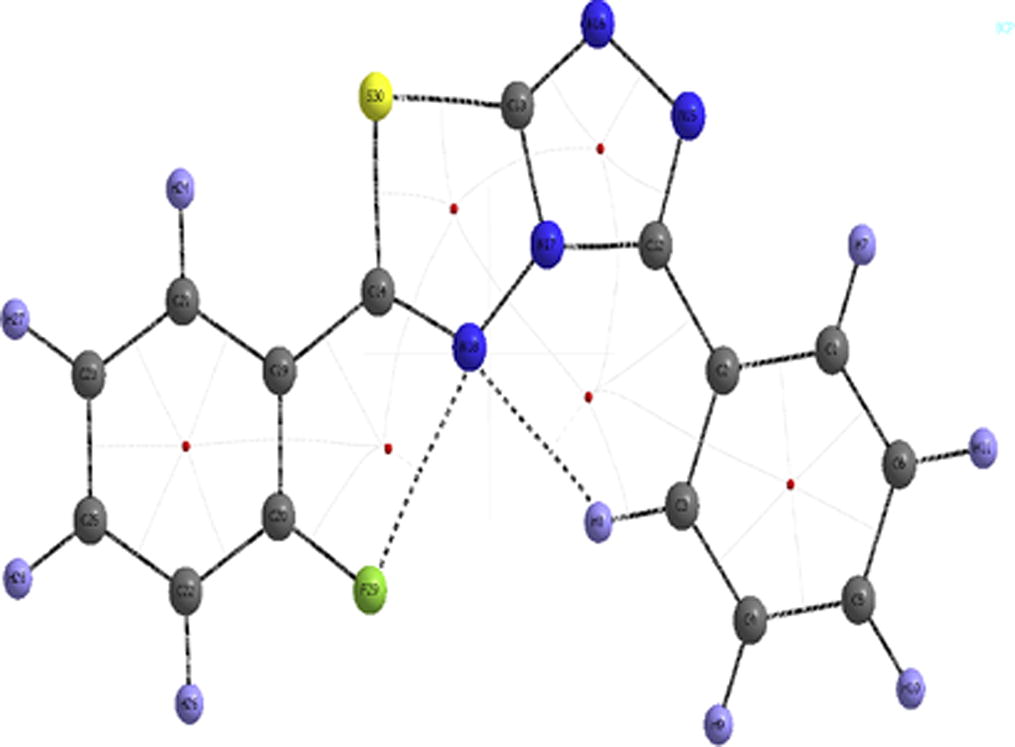
Table 1 Topological parameters for bonds of interacting atoms: electron density (BCP), Laplacian of electron density (
BCP), kinetic electron energy density (G BCP), potential electron energy density (VBCP), total electron energy density (HBCP), estimated interaction energy (Eint) at bond critical point (BCP).
3.2 Electronic property and HOMO-LUMO surfaces
The HOMO represents the ability to donate an electron by an atom while LUMO represents the ability to accept. The HOMO and LUMO energies are calculated by DFT/B3LYP method. The electronic parameters, such as highest occupied molecular orbital (HOMO) energy (E-HOMO), lowest unoccupied molecular orbital (LUMO) energy (E-LUMO) and band gap energy (ΔE = FLUMO-FHOMO) are described theoretically [Citation39]. The energy (Egap) between HOMO and LUMO in the present study is found to be 3.71 eV. In this molecule LUMO is located over whole molecule however HOMO is located only on benzene ring. The transition from HOMO → LUMO in the molecule indicates charge transfer from benzene ring to thiazole ring. The molecular electrostatic potential (MESP) is a map of potential i.e. electrostatic potential over constant electron density of all molecules. It displays molecular size and shape as well as positive, negative, and neutral electrostatic potential regions in terms of color grading scheme. In this color grading schemes, the blue color represents the most electropositivity i.e. low electron region, whereas the red color corresponds to the most electronegative center or electron rich regions [Citation40,Citation41] .The MESP plot of title molecule is display in . From this fig. ring R2 is covered by red region so it is the most electronegative region of title molecule. On the basis of energy of frontier orbitals, HOMO,
LUMO [energy of highest occupied molecular orbital, Energy of lowest unoccupied molecular orbital respectively] the different global reactivity descriptors such as electronegativity
(tendency to attract electrons), chemical potential
(energy that can be absorbed or released), global hardness
(hardness basically signifies the resistance against the deformation or polarization of the electron cloud of the atoms, ions or molecules under small perturbation of chemical response), global electrophilicity index
(capacity of a species to accept arbitrary number of electrons) and global softness S (Softness signify how easily a molecule becomes polarized) are computed using equations as given below [Citation42] ().
Fig. 3 MESP surfaces of thiazol.

Fig. 4 HOMO, LUMO surfaces of thiazol.
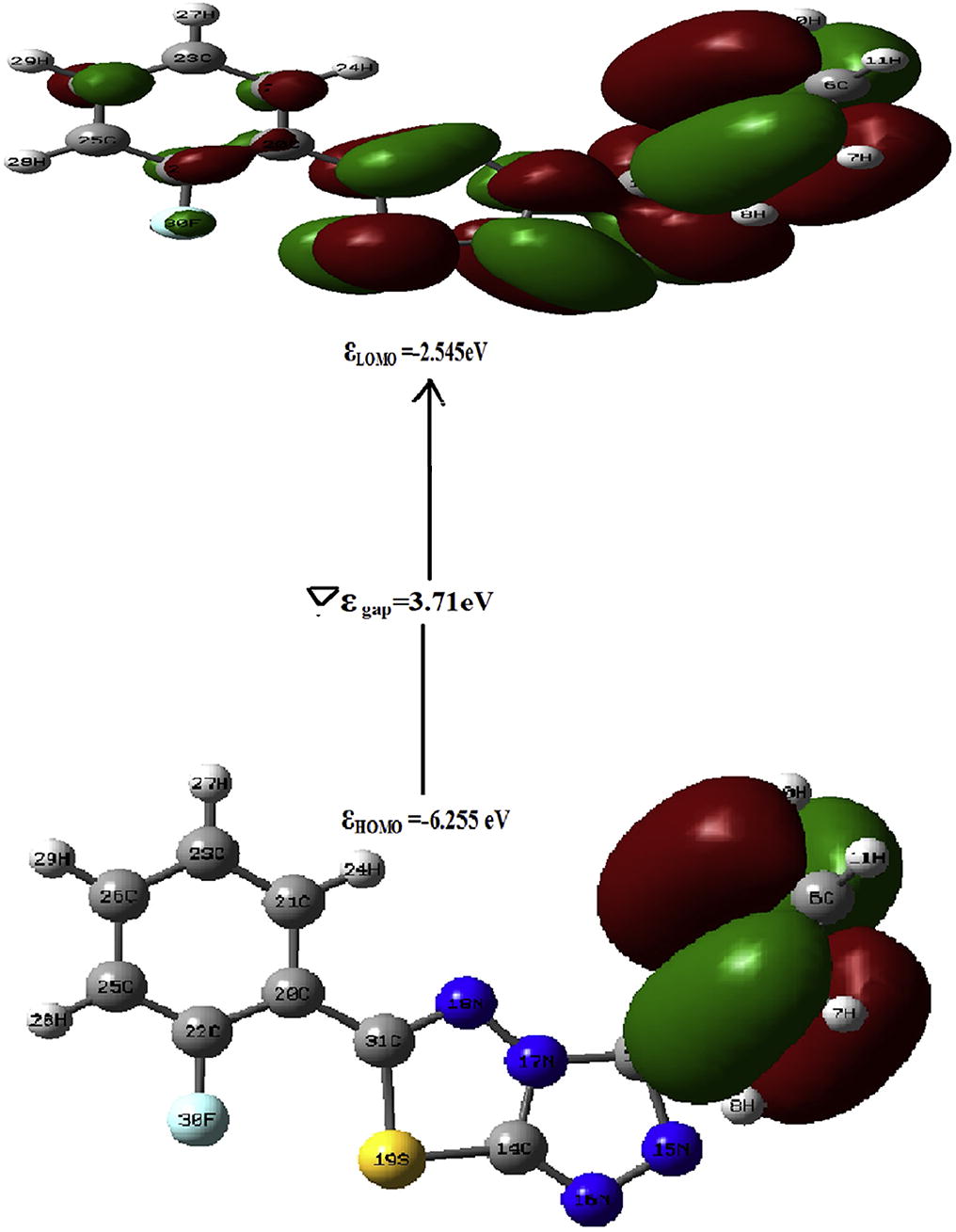
The energies of frontier molecular orbitals (HOMO,
LUMO), energy band gap (
LUMO–
HOMO), electronegativity (
), chemical potential (
), global hardness (
), global softness (S), and global electrophilicity index (
) of the molecule have been listed in . On the basis of
HOMO and
LUMO, these parameters are calculated by using equation given below [Citation43Citation[44]–Citation45]
Table 2 Electronic parameters of title compound calculated by DFT/B3LYP method and 6-311G(d, p).
3.2.1 Fukui function
The Fukui function (FF) [Citation46,Citation47] of a molecule provides information about reactive sites and the method of understanding as well as categorizing chemical reactions. The atom with the highest FF value is highly reactive site as compared to the other atoms in the molecule. The use of the Fukui functions for the site selectivity in title molecule for nucleophilic and electrophilic attacks has been made with special emphasis on the dependence of the Fukui values with the basis of B3LYP/6-311G(d, p) level of theory. Using the Mullikan atomic charges of neutral, cation, and anion, state of the molecule, the Fukui functions (,
,
), local softness (
,
,
) [Citation48] and local electrophilicity indices (
,
,
) are calculated using the following equations
where
is electronic population of atom k in the neutral molecule with N electrons.
Local softness and electrophilicity indices are calculated using
where +, −, and 0 signs show nucleophilics, electrophilics, and radical attacks, respectively. The Fukui functions, for all the carbon atoms in both benzene ring (R1 R4) and sulfar atom in thiazole ring (R3), calculated by using combination of DFT/B3LYP method and 6-311G (d, p) basis set are listed in . From , it can be seen that the carbon of both benzene ring, 14C and 31C are more reactive sites for nucleophilic and 26C as an electrophilic reactive attacks. However 30S atom of the thiazole ring shows more favorable site for both electrophilic and nucleophilic substitution than ring carbon atoms. Furthermore, 30S site is more reactive to the electrophilic than nucleophilic substitution because
.
Table 3 Fukui values on the basis theory of the Mulliken atomic charges of neutral, cation, and anion, the Fukui functions (,
,
).
Time dependent density functional theory (TD–DFT) method is important tool for studying the nature of the transitions of UV–vis spectrum of the compound. Optimized geometry is obtained for TD-DFT calculation by using same level theory. UV spectra of the molecule, calculated by combination of DFT/B3LYP method and 6-311G (d, p) basis set is shown in . In this graph, two prominent peaks observed at 4.02 eV (294 nm), 2.03 eV (610 nm) originate mainly due to H → L (37%), H-4 → L (43%) transitions respectively. These transitions are shown in and listed in . On the basis of the calculated molecular orbital coefficients analyses electronic transition are assigned to n → * and π → π* respectively.
Fig 5 Electronic Transition of TD-DFT of title molecule by DFT/B3LYP method.
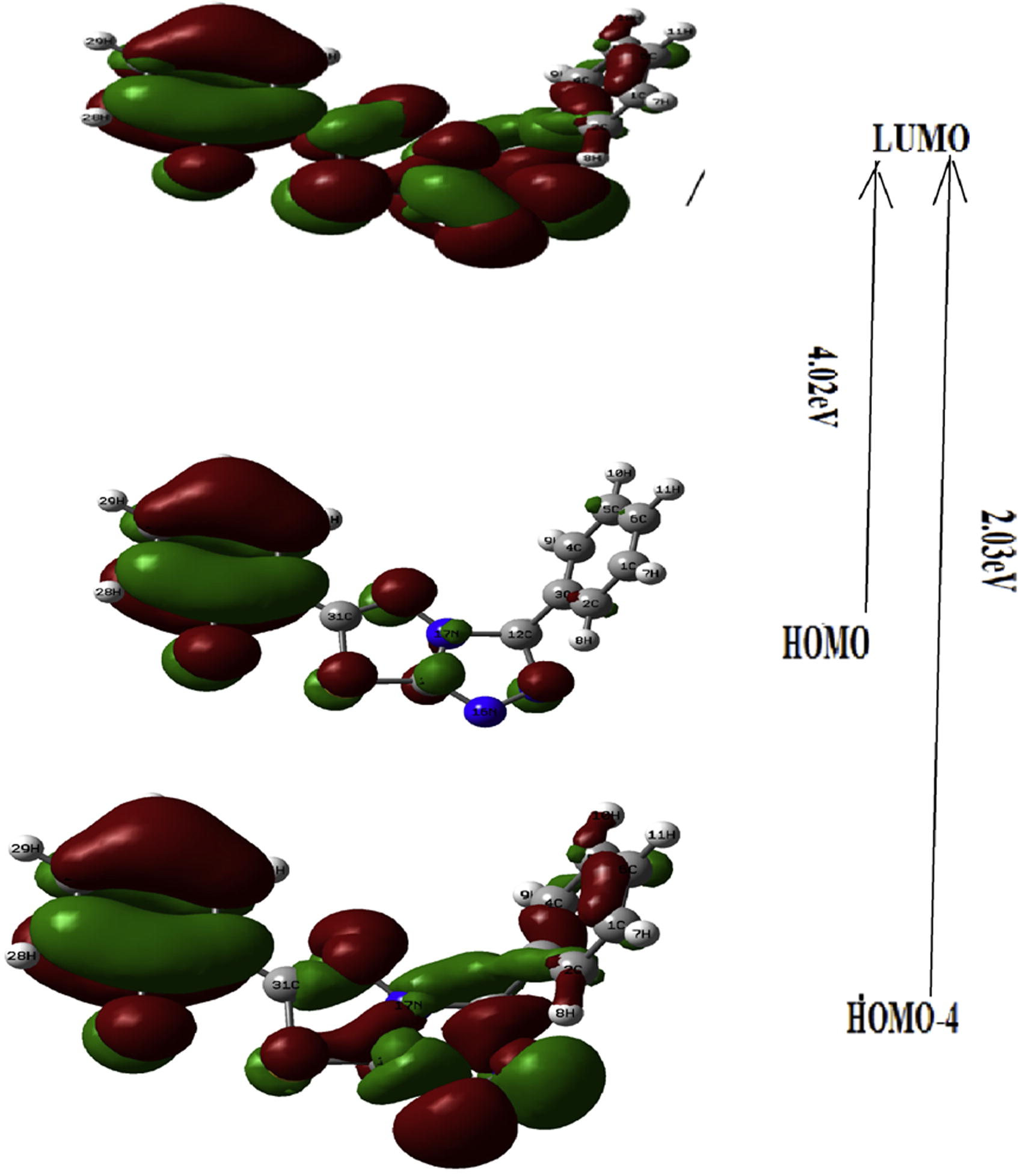
Fig 6 UV spectra of title molecule calculated by combination of DFT/B3LYP method and 6-311G(d,p) basis set.
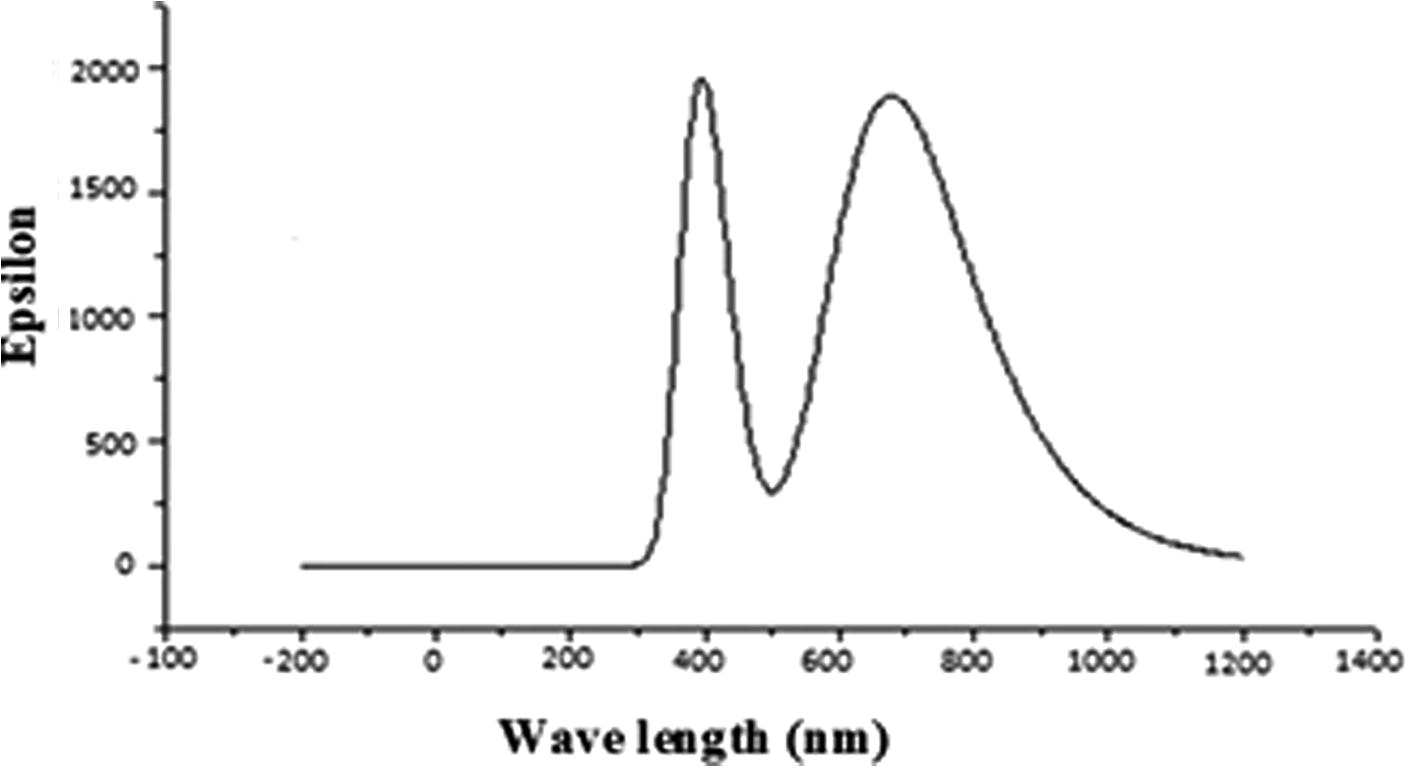
Table 4 The observed UV–vis spectra as calculated by TD-DFT method at B3LYP/6-311 G (d, p) level.
3.3 Non linear optical parameter
The Gaussian 03 program is used to calculate the dipole moment () and polarizability (
) of the molecules, based on the finite field approach. The first hyperpolarizability (
) and polarizibility of the title molecule are calculated by using same level theory and listed in . In the presence of applied electric field, energy is function of applied electric field. Hyperpolarizability and Polarizability are described as the response of a system due to application of the electric field [Citation49]. The Hyperpolarizability and Polarizability are determined by nonlinear optical properties (N L O) of the system as well as strength of molecular interaction [Citation50,Citation51] . First order Hyperpolarizability of the system is defined by 3 × 3 × 3 matrix of is a third rank tensor. The third rank matrix which contains 27 components can be reduced to 10 components by using Kleinman symmetry [Citation52]. All components of
are coefficients in the Taylor series expansion of the energy. When weak and homogeneous external electric field is applied, this expansion becomes
where
is the energy of the unperturbed molecules,
the field at the origin
,
, and
are the components of dipole moment, polarizability and the first hyperpolarizabilities respectively.
Table 5 Polarizability and Hyper Polarizability of title compound calculated by same level theory.
The total dipole moment and the mean polarizability and hyperpolarizability using x,y,z components are defined as
where
,
,
are X,Y,Z components of dipole moment
where
,
,
are the components of polarizibility in XX, YY, ZZ planes
where
are components of hyperpolarizibility along a direction and in bc plane and (1 a.u. = 3.6998 × 10−30 e.s.u.).
The calculated value of dipole moment is 6.29 D. In title molecule and
have greater contribution of polarizibility which indicates that the molecule is more polarized along Y and X directions. The calculated value of hyperpolarizibily of given molecule is nearly same as, 2-{[5-(adamantan-1-yl)-4-methyl-4H-1,2,4-triazol-3-yl]sulfanyl}-N,N-dimethylethanamine [Citation53], and nearly one fifth of Thiazole Azo Dyes.[Citation54] Urea is benchmark compound for NLO properties, therefore we compare our results with urea. The calculated value of hyperpolarizibily of the compound is nearly nine times greater than hyperpolarizibily of urea so it is a good claimant as a NLO material.
3.4 Vibrational analysis
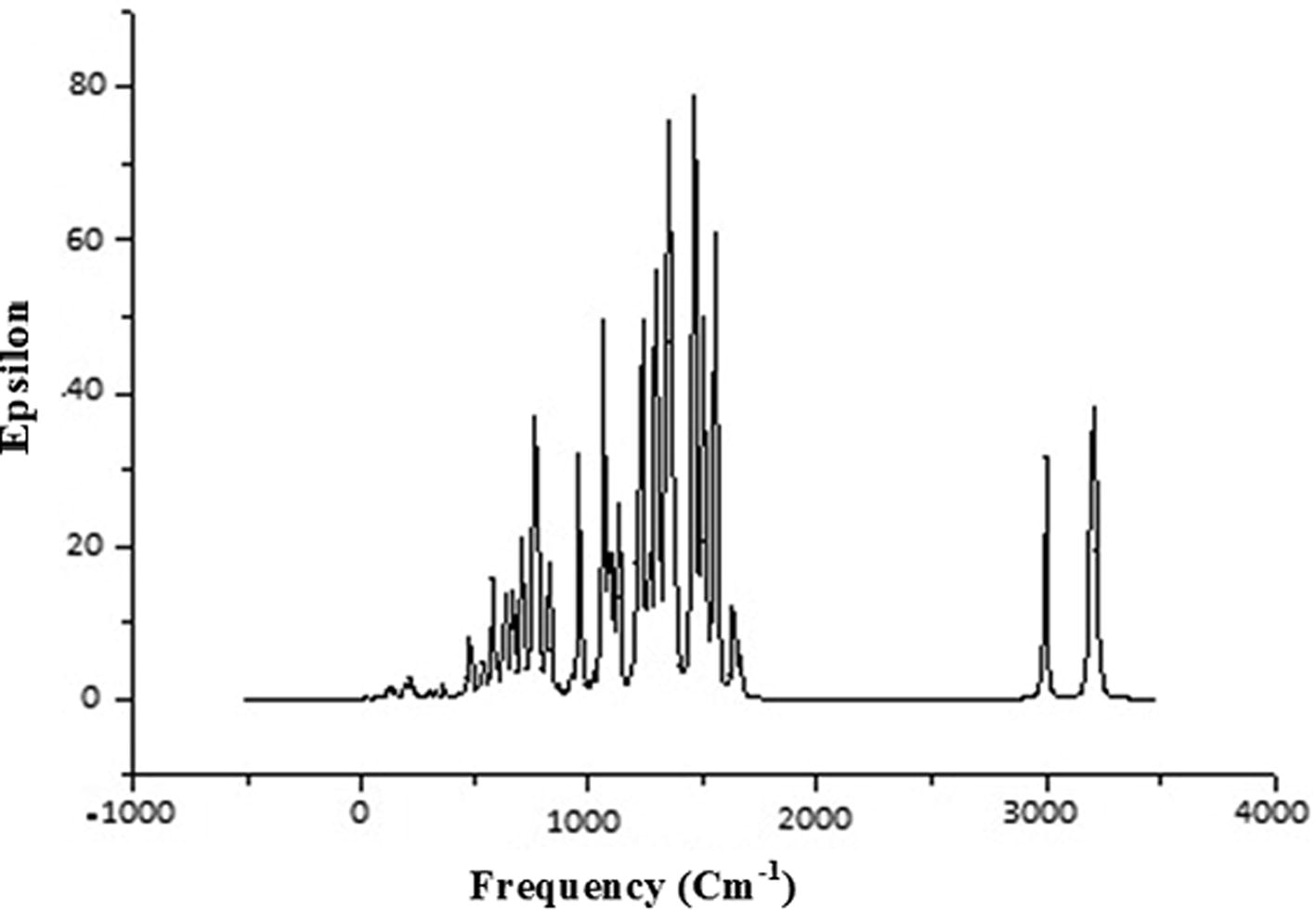
The molecule under study contains 30 atoms, so there are 3N-6 modes of vibrations. All mode of vibration are studied with the help of combination of Gauss View and VEDA 4.0 program package [Citation55]. In our calculations we ignore electron- electron correlation as well as an-harmonicity [Citation56,Citation57] . These approximations cause calculated frequencies to be in higher region than experimental results, therefore we scale calculated frequency by 0.9688. [Citation58] Some of selected modes of vibrations are listed in and calculated IR spectra of the molecule is also plotted in , using same level theory. Some important modes of vibrations are discussed below.
Table 6 Theoretical (selected) vibrational wave numbers of title molecule using B3LYP/6–311G (d, p) and their assignment.
Fig 7 Calculated IR spectra of title molecule by using DFT/B3LYP method.
3.4.1 C-H stretching
The hetero aromatic structure shows the presence of C–H stretching vibrations in the region 2800–3100 cm−1 [Citation58]. In the present study the C–H stretching vibration of the title compound is observed at 3066–3108 cm−1. Two sharp back to back peaks observed at 3077 cm−1, 3076 cm−1 due to (C-H)R1 and mixing of
(C-H)R1,R4 with PED 24%, 43% respectively, are well matched with experimental as well as calculated IR spectra of 2-(4-methoxyphenyl)benzo[d]thiazole at 3076 cm−1 [Citation59]. An intense mixing of modes are observed at 1625 cm−1 due to
(CH) R1 with PED 16%. In mid region of spectra very intense and sharp polarized peaks are observed at 1611 cm−1 and 1605 cm−1 due to
(CH)R1,
(CH)R4 respectively with PED 47% and 76%. In lower region some out of plane bending and twisting mode of vibrations occur.
3.4.2 C-N vibrations
The determination of C-N vibrations is a difficult task, because of the mixing of vibration in this region. Silverstein [Citation58] has first time assigned C-N stretching absorption in the range of 1382–1266 cm−1. The C-N stretching vibration (15N-12C) and (17N-12C) in the molecule observed at 1266 cm−1 and 1263 cm−1 with PED contribution 15% and 46% respectively, are well matched (1267 cm−1) with C-N stretching mode and lies in the same region as 2-(4-methoxyphenyl)benzo[d]thiazole [Citation59]. In lower rage of frequency some other in plane bending vibration modes (14C-18N-17N) and torsion (14C-18N-17N-12C) are obtained at 1088 cm−1 and 654 cm−1 respectively with PED contribution 20% and 79%.
3.4.3 C-C vibrations
Normally the (C-C) stretching vibrations are expected within the middle region of spectra. The C–C stretching vibrations of the aromatic compounds, calculated by B3LYP/6-311G (d, p) method, are at 1600 cm−1, 1586 cm−1, 1568 cm−1, 1500 cm−1 and 1293 cm−1 in thiazole ring however in benzene ring C–C aromatic stretching mode of vibrations lies in between 1645 and 847 cm−1. The other modes of vibration i.e. in-plane and out-of-plane bending are also observed at lower frequencies.
3.4.4 Other modes of vibration
Some other modes of vibrations are also reported at lower range of frequencies. In thiazole ring intense plane polarized mode of vibration due to (29F-20C) with PED 19% is obtained at 1207 cm−1. In this study below 1000 cm−1 half segment of thiazole ring experiences torsion
(14C-17N-18N-30S) at 936 cm−1 with PED 21%, however opposite half segment of thiazole ring experiences very intense peak of torsion τ (29F-19C-22C-20C) at 520 cm−1 with PED 27%. In fuoro ring a medium intense polarized mode of vibration with polarization vector along the plane of ring, due to
(29F-20C), is obtained at 1207 cm−1 with PED 15%. Some other modes of vibrations are reported in table however those mode of vibration are not reported for which PED < 15%.
3.5 Donor acceptor interaction
3.5.1 NBO analysis
NBO gives the most suitable ‘natural Lewis structure’ because all orbitals include the highest possible percentage of the electron density (ED). NBOs deliver an accurate method for studying intra and intermolecular interaction as well as charge transfer or conjugative interaction in different molecular systems [Citation60]. The value of E(2) shows the strength of interaction in between electron donors and electron acceptors, i.e., As the value of E(2) increases the strength of interaction increases and vice-versa. For each donor NBO(i) and acceptor NBO(j), the strength of delocalization interaction (or stabilization energy) E(2) associated with electron delocalization between donor and acceptor is estimated by the second order energy lowering as [Citation61,Citation62]
where
is the population of donor orbital or donor orbital occupancy;
,
are orbital energies (diagonal elements) of donor and acceptor NBO orbitals respectively; Fij is the off–diagonal Fock or Kohn–Sham matrix element between i and j NBO orbitals. The second-order perturbation analysis of Fock matrix in NBO basis are listed in . From this table we see that strong intra-molecular hyper conjugative interactions are formed by orbital overlap Lp(1)N15/N17,Lp(2)S30,
(C-C),
(C-H)π(C-C)
*(C-C),
*(C-H),
*(C-C) which results in intra-molecular charge transfer causing stabilization of the system. From , interactions between C1-C2 to C3-C4, C5-C6 of
(C1-C2) →
*(C3-C4) and
(C1-C2) →
*(C3-C4) with electron density 0.9897 e stabilize by 10.28 kcal/mol, 10.49 kcal/mol respectively. The interaction between C3-C4 to C1-C2, C4-C5, C12-C17 of
(C3-C4) →
*(C1-C2)
(C3-C4 →
*(C5-C4),
(C3-C4) →
*(C12-C17) with electron density 0.833e stabilizes by 10.04 kcal/mol, 9.72 kcal/mol and 3.11 kcal/mole respectively. Similarly interaction between C5-C6 to C3-C4, C1-C2, of
(C5-C6) →
*(C3-C4)
(C5-C6) →
*(C1-C2) with electron density 0.829 e stabilizes by 9.6 kcal/mol, 10.54 kcal/mol respectively. A strong interaction observed in between lone pair LP(1)N17 →
*(C14-N18),
*(N18-C31) with energy 21.09 kcal/mol and 20.25 kcal/mol respectively are comparable with interaction has been observed due to the electron density transfer from the lone pair (LP1) of nitrogen atom to antibonding orbitals
*(C–N) and
*(N–C) with stabilization energies 41.07 and 46.78 kcal/mol, respectively in similar molecular system 2-{[5-(adamantan-1-yl)-4-methyl-4H-1,2,4-triazol-3-yl]sulfanyl}-N,N-dimethylethanamine [Citation53]. Since
orbitals have lower occupancies than σ, correspondingly showing more electron- donating ability in comparison to
orbital. This also shows that [LP(1) (S30) →
*(C14-C16)] is the most intensive interaction between the acceptor and donor which results in the molecular stability. A very strong interaction by
* to
* orbital overlap of
*(C22-C25) →
*(C23-C26) stabilizes by 153.69 kcal/mol. In this system stabilization occurs by intermolecular interactions of orbital overlap among
and
* orbitals, which results in intermolecular charge transfer (ICT). Since ICT makes molecule more polarized, it must be responsible for the NLO properties of molecule. Consequently the compound may be used for NLO materials in future.
Table 7 Second–order perturbation theory analysis of the Fock matrix, in the NBO basis for intermolecular interactions within title molecule: Stabilization energy of interactions (E(2)), Energy difference between donor (i) and acceptor (j) NBO orbitals (Ej–Ei), Fock matrix element between i and j NBO orbitals (Fij).
3.6 Biological properties
Before discussing the biological activity of the molecule aqueous solubility (log S) and Lipophilicity (log P) are calculated. For evaluating log P and log S, ALOGPS 2.1 program [Citation63] is used. This program developed by Tetko et al. [Citation64Citation[65]–Citation66] is based on electrotopological state indices and associative neural network modeling. The Structure-property relationship studies are justified by these two parameters. The transport property of drugs and their interaction with receptors are closely related by Log P however bioavailability is closely related by log S. The calculated value of log P is 2.34 which suggests that molecule is able to diffuse across the cell membranes, so it can be utilized in pharmacological applications. In general 85% of drugs have log S values in between the range of −1 to −5.36. The calculated value of log S (−3.30) further approves the permeability of molecules through cell membranes. Some biological activities of title molecule are calculated with the help of PASS software. PASS calculates 900 pharmacological properties, molecular mechanisms of action, mutagenicity, carcinogenicity, teratogenicity, and embryotoxicity. PASS predicts these properties on the basis of structure activity relationships for the training set, considering more than 46 000 drugs, drug candidates, and prime compounds whose biological activity are resolute experimentally. The calculated results by PASS software have average accuracy of about 85% [Citation67]. In this study we only listed those biological activities, in , for which Pa > 70%. The molecule shows high value of biological activity for Chloride peroxidase inhibitor. The Chloride peroxidase belongs to the family of enzymes, which catalyzes the chlorination of organic compounds. It employs one cofactor, which may be either heme or vanadium [Citation68]. The molecule inhibits chlorination of organic compounds in blood stream which prevents bacterial disease. It is used for Phobic disorders treatment also. Phobias are the most common type of anxiety disorder, in which a person experiences an extreme or irrational fear of a place, object, animal, or situation. Corticosteroid side-chain isomers exist in all organs but show highest activity in liver and kidneys. The molecule inhibits Corticosteroid side-chain isomers in liver and kidneys and protect these organ from multiple myeloma. Glucocorticoids (0.646) are part of the response mechanism in the immune system which decreases certain features of immune function, such as drop of inflammation [Citation69]. So they are used as medicines to treat diseases caused by an intense resistant system. They also inhibit growth in cancer cells, therefore high doses of Glucocorticoids are used to treat cancer. This contains inhibitory effects on lymphocyte production, as in the handling of lymphomas and the moderation of side effects of anticancer drugs. Designing new anti-inflammatory in Myotonic dystrophy diseases, agents require the identification of targets, which when inhibited can kill the affected cells. We have done molecular docking studies by using Swiss-Dock web server [Citation70]. MBNL1 receptor is identified as an efficient target, based on the prediction by Swiss-Dock web server. Muscleblind-like (Drosophila), also known as MBNL1, is a protein which is encoded in humans by the MBNL1 gene [Citation71Citation[72]–Citation73] . MBNL1 is stated in the early heart, and its levels rise across fatal and postpartum development. The docking score is expected to have minimum interaction energy value (e-value). As more negative the e-value is the docking becomes more efficient. Docking process does not cover on a specific region but over entire protein. The docking picture of the molecule, obtained from the UCSF chimera software, is shown in . The Full Fitness score for the title molecule is −541.58 a.u. and binding affinity (ΔG in kcal/mol) −5.46 a.u., which suggest that it has good binding affinity. The results obtained from docking studies suggest that the compound can be used as an anti-inflammatory agent in Myotonic dystrophy.
Fig 8 Molecular Docking of MBNL1 protein with title molecule.
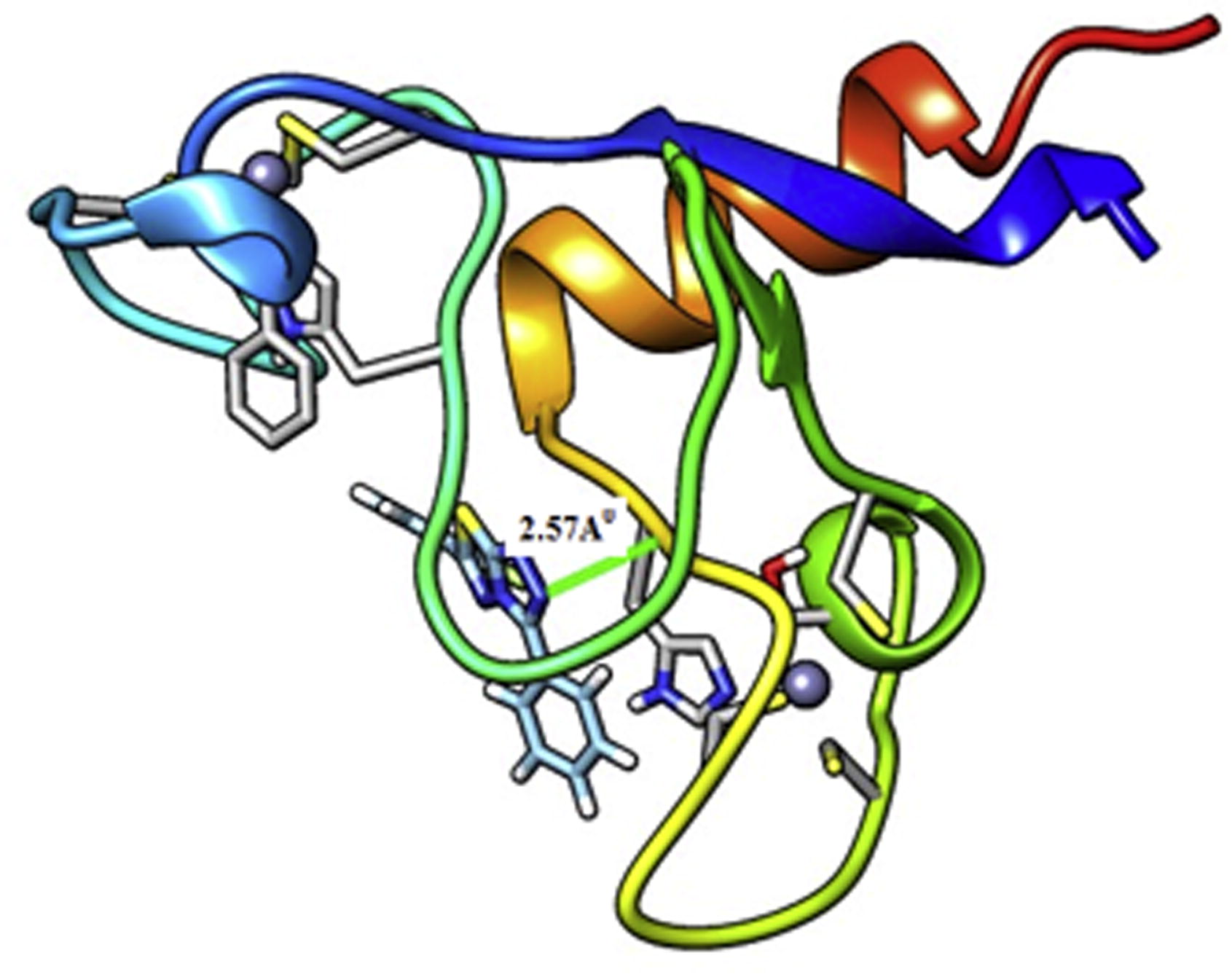
Table 8 Some biological activities calculated by PASS with Pa > 70%.
4 Conclusion
Equilibrium geometries, harmonic frequencies, electronic properties, NBO, NLO analysis, TDDFT and biological activity of title molecule are determined and analyzed using combination of DFT/B3LYP level of theory using 6-311G (d, p) basis set. The theoretical calculation of UV–vis spectra shows a medium intense peak (at 610 nm) corresponding to H-4 → L. The NBO analysis shows that strongest interaction with energy 153.69 kcal/mol corresponding to *(C22-C25) →
*(C23-C26) stabilizes flouro ring. The chemical reactivity of the molecule is explained by energy gap (3.71 eV) and plot of HOMO-LUMO. FF shows that 14C and 31C are better nucleophilic centers however 30S shows better electrophilic center. Full Fitness score corresponding to docking of the molecule with and MBNL1 protein is −541.58 a.u. The molecular docking of title molecule suggests that it can bind and inhibit the receptor enzymes.
Supplementary data 1
Download MS Word (28.5 KB)References
- J.A.ZoltewiczL.W.DeadyQuaternization of heteroaromatic compounds: quantitative aspectsAdv Heterocycl Chem221197871121
- T.EicherS.HauptmannThe chemistry of heterocycles: structure, reactions, syntheses, and applications2nd ed.2003Theophil Eicher Siegfried Hauptmann
- Jeffrey Y.W.MakWeijunXuDavid P.FairlieThiazoles in peptides and peptidomimeticsW.D.LubellPeptidomimetics-I2017SpringerCham235266
- A.WeissbergerSpecial topics in heterocyclic chemistry1st ed.1977John Wiley
- M.AlajarínJ.CabreraA.PastorP.Sánchez-AndradaD.BautistaHydrazinecarbothioamide group in the synthesis of heterocyclesJ Org Chem71142006150197
- M.KriekF.MartinsR.LeonardiS.A.FairhursD.J.LoweP.L.RoachThiazole synthase from Escherichia coli. An investigation of the substrates and purified proteins required for activity vitroJ Biol Chem2822420071741317423
- A.DondoniP.MerinoDiastereoselective homologation of D-(R)-gylcycereoselective using 2(trimethylsilyl)Thiazole:2-O-benzyle-3,4-Isopropylidene-D-erythroseOrg Synth9521995323326
- E.AmirS.RozenEasy access to the family of thiazole N-oxides using HOF·CH3CNChem Commun21200622622264
- X.ChaiWu.Q.Zhanget al.Design, synthesis and antifungal activities of novel 1,2,4triazole derivatives.Bioorg Med Chem Lett467201131423148
- A.A.El-EmamT.M.IbrahimSynthesis, anti-inflammatory and analgesic activity of certain 3-(1-adamantyl)-4-substituted-5-mercapto-1,2,4-triazole derivativesArzneim-Forsch/Drug Res41199112601264
- A.AboelmagdI.A.I.AliE.M.S.SalemM.Abdel-RazikSynthesis and antifungal activity of some s-mercaptotriazolobenzothiazolyl amino acid derivativesEur. J. Med. Chem.602013503511
- A.A.El-EmamA.M.S.Al-TamimiM.A.Al-OmarK.A.Al-RashoodE.E.HabibSynthesis and antimicrobial activity of novel 5-(1-adamantyl)-2-aminomethyl-4-substituted-1,2,4-triazoline-3-thionesEur J Med Chem68201396102
- Y.LuoS.ZhangZ.J.LiuW.ChenJ.FuQ.F.Zenget al.Synthesis and antimicrobical evaluation of a novel class of 1,3,4-thiadiazole: derivatives bearing 1,2,4-triazolo[1,5- a]pyrimidine moietyEur J Med Chem6420135461
- T.PlechM.WujecU.KosikowskaA.MalmB.KapronStudies on the synthesis and antibacterial activity of 3,6-disubstituted 1,2,4-triazolo[3,4-b]1,3,4-thiadiazolesEur J Med Chem472012580584
- N.F.EweissA.A.BahajajSynthesis of heterocycles. Part VIIJ Heterocycl Chem24198711731181
- Y.KotaiahK.NagarajuN.HarikrishnaC.V.RaoL.Yaminisynthesis, docking and evaluation of antioxidant and antimicrobial activities of novel 1,2,4-triazolo[3,4b][1,3,4]thiadiazol-6-yl)selenopheno[2,3-d]pyrimidinesEur J Med Chem752014195202
- S.N.SwamyPriya B.S.BasappaB.PrabhuswamyK.S.RangappaSynthesis of pharmaceutically important condensed heterocyclic 4,6 disubstituted-1,2,4-triazolo-1,3,4-thiadiazole derivatives as antimicrobialsEur J Med Chem4142006531538
- V.MathewJ.KeshavayyaV.P.VaidyaHeterocyclic system containing bridgehead nitrogen atom: synthesis and pharmacological activities of some substituted 1,2,4-triazolo[3,4-b]-1,3,4-thiadiazolesEur J Med Chem419200610481058
- B.ChaturvediN.TiwariN.NirupamaBassianolide, a New Insecticidal Cyclodepsipeptide from Beauveria bassiana and Verticillium lecaniiAgri Biol Chem423198812291232
- I.KhanS.ZaibA.IbrarN.S.RamaJ.SimpsonCrystalIqbalstructure of 6-(2-fluorophenyl)-3-phenyl-[1,2,4]-triazolo[3,4-b][1,3,4]thiadiazole, C15H9FN4SEur J Med Chem782014167177
- N.ChidanandaB.PoojaryV.SumangalaN.S.KumariP.ShettyT.ArulmoliFacile synthesis, characterization and pharmacological activities of 3,6-disubstituted 1,2,4-triazolo[3,4b][1,3,4]thiadiazoles and 5,6-dihydro-3,6-disubstituted-1,2,4-triazolo[3,4-b][1,3,4]thiadiazolesEur J Med Chem512012124136
- S.Z.AjabshirD.M.SobhanS.N.MasoudSono synthesis and characterization of Ho2O3 nanostructures via a new precipitation way for photocatalytic degradation improvement of erythrosineInt J Hydrogen Energy422220171517815188
- S.Z.AjabshirD.M.SobhanS.N.MasoudGreen synthesis and characterization of Dy2Ce2O7 nanostructures using Ananas comosus with high visible-light photocatalytic activity of organic contaminantsJ Alloy Compd7632018314321
- S.Z.AjabshirD.M.SobhanS.N.MasoudNd2Sn2O7 nanostructures as highly efficient visible light photocatalyst: green synthesis using pomegranate juice and characterizationJ Cleaner Prod19820181118
- S.Z.AjabshirD.M.SobhanS.N.MasoudNd2O3-SiO2 nanocomposites: A simple sonochemical preparation, characterization and photocatalytic activityUltrasonics – Sonochemistry42322018171182
- S.Z.AjabshirD.M.SobhanS.N.MasoudPreparation, characterization and photocatalytic properties of Pr2Ce2O7 nanostructures via a facile wayRSC Adv61062016107785107792
- F.RaziS.Z.AjabshirS.N.MasoudPreparation, characterization and photocatalytic properties of Ag2ZnI4/AgI nanocomposites via a new simple hydrothermal approachJ Mol Liq2252017645651
- A.MonirahHazem A.Al-AlshaikhS.M.Ghabbour MohammedSantiago García-GrandaAbdelbakyAli A.El-EmamCrystal structure of 6-(2-fluorophenyl)-3-phenyl-[1,2,4]-triazolo[3,4-b][1,3,4]thiadiazole, C15H9FN4S ZEITSCHRIFT FÜR KRISTALLOGRAPHIE. Z. Kristallogr.NCS23122016661663
- Gaussian Inc.Gaussian 03 program2004Gaussian Inc.Wallingford
- D.A.PeterssonM.A.AllahamA complete basis set model chemistry. II. Open-shell systems and the total energies of the first-row atomsJ Chem Phys94199160816090
- D.A.PeterssonA.BennettT.G.TensfeldtM.A.AllahamW.A.J.MantzarisA complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elementsJ Chem Phys89198821932218
- Frisch A, Nelson AB, Holder AJ. Gauss view. Pittsburgh, Pa, USA; 2005.
- Keith TA. AIMAll Version 12.09.23 TK Gristmill Software. Overland Park, KS, USA; 2012.
- Glendening ED, Badenhoop JK, Reed AE, Carpenter JE, Weihold F. NBO 3.1 Program Theoretical Chemistry Institute, University of Wisconsin Madison WI; 1996.
- I.F.MattaR.J.BoydAn introduction to the quantum theory of atoms in molecules2007Wiley-VCH VerlagGmbh
- U.KochP.PopelierCharacterization of C-H-O hydrogen bonds on the basis of the charge densityJ Phys Chem A9924199597479754
- I.RozasI.AlkortaJ.ElgueroBehavior of ylides containing N, O, and C atoms as hydrogen bond acceptorsJ Am Chem Soc1224520001115411161
- E.EspinosaE.MolinsC.LecomteHydrogen bond strengths revealed by topological analyses of experimentally observed electron densitiesChem Phys Lett28531998170173
- S.SubashchandraboseH.SaleemY.ErdogduG.RajarajanV.ThanikachalamFT-Raman, FT-IR spectra and total energy distribution of 3-pentyl-2,6-diphenylpiperidin-4-one: DFT methodSpectrochim Acta Part A8212011260269
- J.S.MurrayK.SenMolecular electrostatic potentials1st ed.1996ElsevierAmsterdam
- Scrocco E, Tomasi J, Lowdin (Ed). Methods of molecular quantum mechanics, 2nd ed. New York: Academic Press; 1978.
- R.G.ParrW.YangDensity functional theory of atoms and molecules1989Oxford University PressOxford, New York
- A.F.LeeW.DavidK.J.-A.GallagherA convenient method for the reduction of ozonides to alcohols with borane-dimethyl sulfide complexJ Org Chem546198914301432
- R.G.ParrR.G.PearsonAbsolute hardness: companion parameter to absolute electronegativityJ Am Chem Soc10526198375127516
- R.G.ParrL.SzentpályS.LiuElectrophilicity indexJ Am Chem Soc1219199919221924
- P.K.ChattarajU.SarkarD.R.RoyElectrophilicity indexChem Rev1066200620652091
- A.ValenciaJ.GázquezA.VelaGlobal and local partitioning of the charge transferred in the Parr-Pearson modelJ Phys Chem A12120201740194029
- W.YangW.J.MortierThe use of global and local molecular parameters for the analysis of the gas-phase basicity of aminesJ Am Chem Soc10819198657085711
- C.R.ZhangH.S.ChenG.H.WangStructure and properties of semiconductor Microclusters GanPn(n = 1–4): a first principle studyChem Res Chin U2052004640643
- C.JamesA.RajR.RaghunathanJ.I.HubertV.S.JayakumarStructural conformation and vibrational spectroscopic studies of 2,6-bis(p-N, N-dimethyl benzylidene)cyclohexanone using density functional theoryJ Raman Spectrosc37912200613811392
- J.N.LiuZ.R.ChenS.F.YuanStudy on the prediction of visible absorption maxima of azobenzene compoundsJ Zhejiang Univ Sci B662005584589
- N.B.ColthupL.H.DalyS.E.WiberleyIntroduction to infrared and Raman spectroscopy3rd ed.1990Academic PressBoston, MA
- M.S.AlmutairiA.M.AlanaziE.S.Al-AbdullahA.El-EmamAS.K.PathakR.Srivastavaet al.and FT-Raman spectroscopic signatures, vibrational assignments, NBO, NLO analysis and molecular docking study of 2-{[5-(adamantan-1-yl)-4-0methyl-4H-1,2,4-triazol-3-yl]sulfanyl}-N,N-dimethylethanamineSpectrochimica Acta Part A: Mol Biomol Spectrosc1402015114
- O.I.OsmanDFT study of the structure, reactivity, natural bond orbital and hyperpolarizability of thiazole azo dyesInt J Mol Sci1822017239254
- Jamroz MH, Vibrational energy distribution analysis: VEDA4 program. Warsaw, Poland; 2004.
- J.A.PopleH.B.SchlegelR.KrishnanD.J.DefreesJ.S.BinkleyM.J.Frischet al.Molecular orbital studies of vibrational frequenciesInt J Quant Chem201981269278
- J.A.PopleA.P.ScottM.W.WongL.RadomScaling Factors for Obtaining Fundamental Vibrational Frequencies and Zero-Point Energies from HF/6–31G* and MP2/6–31G* Harmonic FrequenciesIsr J Chem331993345350
- V.KrishnakumarJ.R.XavierMolecular and vibrational structure of 2-mercapto pyrimidine and 2,4-diamino-6-hydroxy-5-nitroso pyrimidine: FT-IR, FT-Raman and quantum chemical calculationsSpectrochim Acta, Part A6322006454463
- H.ArslanÖ.AlgülSynthesis and Ab Initio/DFT Studies on 2-(4methoxyphenyl) benzo[d]thiazoleInt J Mol Sci882007760776
- Y.ErdogduO.UnsalanM.T.GulluogluFT-Raman, FT-IR spectral and DFT studies on 6, 8- dichloroflavone and 6, 8-dibromoflavoneJ Raman Spectrosc412010820828
- Y.ErdogduO.UnsalanM.AmalanathanJ.I.HubertInfrared and Raman spectra, vibrational assignment, NBO analysis and DFT calculations of 6-aminoflavoneJ Mol Struct980320102430
- N.GonoheH.AbeN.MikamiM.ItoTwo-color photoionization of van der Waals complexes of fluorobenzene and hydrogen-bonded complexes of phenol in supersonic jetsJ Phys Chem8917198536423648
- www.vcclab.org.
- J.J.HuuskonenD.J.LivingstoneI.V.TetkoNeural network modeling for estimation of partition coefficient based on atom-type electrotopological state indicesJ Chem Inf Comput Sci4042000947955
- I.V.TetkV.Y.TanchukT.A.KashevaA.E.VillaEstimation of aqueous solubility of chemical compounds using E-state indicesJ Chem Inf Comput Sci41200114881493
- I.V.TetkV.Y.TanchukT.A.KashevaA.E.VillaPrediction of n-octanol/water partition coefficients from PHYSPROP database using artificial neural networks and E-state indicesJ Chem Inf Comput Sci41200114071421
- W.L.JorgensenE.M.DuffyPrediction of drug solubility from Monte Carlo simulationsBioorg Med Chem Lett1011200011551158
- A.SadymA.LaguninD.FilimonovV.PoroikovPrediction of biological activity spectra via the InternetSAR QSAR Environ Res145–62003339347
- A.ButlerJayme N.Carter-FranklinThe role of vanadium bromoperoxidase in the biosynthesis of halogenated marine natural productsNat Prod Rep2112004180188
- www.swissdock.ch/.
- K.IshikawaT.NagaseD.NakajimaN.SekiM.OhiraN.Miyajimaet al.Prediction of the coding sequences of unidentified human genes. VIII. 78 new cDNA clones from brain which code for large proteins in vitroDNA Res451997307313
- J.W.MillerC.R.UrbinatiP.Teng-UmnuayM.G.StenbergB.J.ByrneRecruitment of human muscle blind proteins to (CUG)(n) expansions associated with myotonic dystrophyEMBO J1917200044394448
- T.H.HoB.N.CharletM.G.PoulosG.SinghM.S.SwansonT.A.CooperMuscle blind proteins regulate alternative splicingEMBO J2315200431033112
Appendix A
Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ejbas.2018.10.001.