
Abstract
Y/MCM-48 composite molecular sieve was hydrothermally synthesized at different crystallization temperatures and crystallization times using ethyl orthosilicate as Si source and cetyltrimethyl ammonium bromide as template with the aid of fluoride ions and was characterized by X-ray diffraction, N2 physical adsorption technique, scanning electron microscopy and transmission electron microscopy. The thermal, hydrothermal, acidic, and basic stabilities of the Y/MCM-48 composite were investigated. The results show that Y/MCM-48 composite molecular sieve with meso- and microporous structures was synthesized successfully at 120 °C for 36 h. The Y/MCM-48 composite has the surface area of 864 m2/g and the average pore size is ca. 2.48 nm. The bi-porous structure in composite molecular sieve still maintains its stability even after thermal treatment at 800 °C for 4 h or hydrothermal treatment at 100 °C for 48 h. After treatment in 1 mol/L hydrochloric acid solution or 1 mol/L sodium hydroxide solution for 48 h, the Y/MCM-48 composite exhibits good acidic stability. The acidic stability is superior to the basic stability at the same treatment time.
1 Introduction
It is well known that the microporous molecular sieves were widely used in the petrochemical industry owing to their several advantages such as good thermal and hydrothermal stabilities, high surface area, uniform pore channel, and high acidity [Citation1–Citation4]. However, these microporous molecular sieves possess small pore size (<2 nm), which severely limited their applications in macromolecule catalysis reaction [Citation5]. M41S mesoporous molecular sieves, discovered in the early 1990s [Citation6], have attracted considerable attention in some areas such as macromolecular reaction, petrochemical industry, adsorption, and catalysis due to their unique pore structure, high surface area, and adjustable pore size ranging from 1.5 to 10 nm [Citation7–Citation12]. Much effort showed that mesoporous molecular sieves exhibited good catalytic behavior in many reactions such as cracking, isomerization, alkylation, hydrogenation, hydroxylation, and carbonylation, etc. However, mesoporous molecular sieves have some disadvantages such as neutral framework, weak surface acidity, and poor hydrothermal stability, which limited greatly their application in industry and catalysis.
In order to overcome the limitation of single microporous or mesoporous molecular sieves and to combine the advantages of each pore size regime, a new type molecular sieve which combines composite molecular sieve with meso-microporous structure was expected to provide attractive features. Therefore, increasing interest has been sparked in synthesis and application of composite molecular sieve to the combination of performance of meso- and microporous molecular sieves [Citation13–Citation16]. Owing to their strong acidity and high hydrothermal stability, composite molecular sieves with bi-pore structure have some potential applications in fine chemical and petrochemical industries [Citation17,Citation18]. Recently, several routes have been adopted to synthesize composite molecular sieve with meso- and microporous frameworks [Citation19Citation[20]–Citation22]. Most of the previous reports aimed at the synthesis and application of composite molecular sieves based on MCM-41 mesoporous molecular sieve [Citation23Citation[24]–Citation25]. Little attention is paid at the investigation of composite molecular sieve based on MCM-48 molecular sieve. Compared with the one-dimensional channels of MCM-41, MCM-48 mesoporous molecular sieve has an attractive and unique cubic arrangement of three-dimensional interwoven structure [Citation26], which can effectively avoid pore block by guest molecular and increase the diffusional speed of reactants in the pores, and hence the cubic three-dimensional MCM-48 is superior to one-dimensional MCM-41 in the fields of catalysis, adsorption and separation reaction of macromolecule [Citation27].
In this paper, we combined the advantages of meso- and microporous molecular sieves to synthesize the Y/MCM-48 composite molecular sieve by hydrothermal method with the aid of fluoride ions. In particular, we focus on investigation on thermal, hydrothermal, acidic and basic stabilities of Y/MCM-48 composite molecular sieve. Various methods such as XRD, SEM, TEM and N2 physical adsorption technique were used to characterize the resulting Y/MCM-48 composite molecular sieve. Moreover, the effect of crystallization temperature and time on synthesis process was investigated in detail.
2 Experimental
2.1 Materials
The chemicals used for the synthesis of the composite molecular sieve were tetraethyl orthosilicate (TEOS), cetyltrimethyl ammonium bromide (CTAB), sodium hydroxide (NaOH) and sodium fluoride (NaF). They were purchased from Shanghai Chemical Reagent Corporation, PR China. All reagents were of analytical grade. Y type molecular sieve (NaY, Si/Al = 2.4) was purchased from the Catalyst Work of Nankai University, China.
2.2 Synthesis of composite molecular sieve
Synthesis of the composite molecular sieve was carried out by hydrothermal method. According to the molar ratio of 1TEOS:0.65CTAB:0.5NaOH:62H2O:0.1NaF in starting material, a typical preparation procedure was carried out as follows. Firstly, 0.9 g of NaOH and 0.19 g of NaF were dissolved in 50 ml distilled water, and then 10.62 g of CTAB was added into the mixture under vigorous stirring at 35 °C for 50 min, and 10 ml of TEOS was slowly added into the above mixture. After stirring for another 1 h, the gelatinous mesoporous precursor was obtained. After that, 7.1 g of Y type molecular sieve was added into the above gelatinous mesoporous precursor with stirring for additional 1 h. The mixture was transferred to a 100 ml Teflon-lined autoclave and crystallized at 100, 120, 140 and 160 °C for 24, 36, 48 and 72 h in an oven, respectively. After cooling to room temperature, the precipitate was filtered, washed with deionized water and dried at 60 °C for overnight in an oven. The dried samples were heated to 550 °C at a heating rate of 2 °C/min and kept at 550 °C for 6 h, denoted as Y/MCM-48.
2.3 Stability test
2.3.1 Thermal and hydrothermal stabilities evaluation
For hydrothermal stability test, 0.3 g of the Y/MCM-48 sample was added into a 100 ml Teflon-lined stainless autoclave containing 80 ml of H2O and hydrothermally treated at 100 °C for 12, 24, 48 and 72 h, designated as Y/MCM-48-12, Y/MCM-48-24, Y/MCM-48-48 and Y/MCM-48-72, respectively. For thermal stability test, 0.3 g of the Y/MCM-48 sample was calcined at 700, 800 and 900 °C for 4 h, respectively, marked as Y/MCM-48-700, Y/MCM-48-800 and Y/MCM-48-900.
For comparison, the thermal and hydrothermal stabilities of the pure silica MCM-48 and NaY zeolite were checked by employing the same method as described above.
2.3.2 Stability of the Y/MCM-48 composite in acidic and basic solution
The acid stability test, 0.3 g of the Y/MCM-48 sample was dispersed into 50 ml 1 mol/L HCl solution with stirring at ambient temperature and was treated for 12, 24 and 48 h, respectively, denoted as Y/MCM-48-12(A), Y/MCM-48-24(A) and Y/MCM-48-48(A). For the basic stability test, 0.3 g of the Y/MCM-48 sample was dispersed into 50 ml 1 mol/L NaOH solution with stirring at room temperature and was treated for 12, 24 and 48 h, named as Y/MCM-48-12(B), Y/MCM-48-24(B) and Y/MCM-48-48(B), where A and B stand for acid and base, correspondingly.
2.4 Characterization
The XRD patterns of samples were recorded on a Rigaku D/MAX 2500PC powder X-ray diffraction instrument with Cu Kα radiation (λ = 0.15418 nm). The measurement conditions of XRD at low angle: 40 kV, 50 mA and the scanning range 2θ = 1–10°; the measurement conditions of XRD at high angle: 40 kV, 200 mA and the scanning range 2θ = 4–40°. Specific surface area and pore size were measured by using a NOVA2000e analytical system made by Quantachrome Corporation (USA). The specific surface area was calculated by Brunauer–Emmett–Teller (BET) method. Pore size distribution and pore volume were calculated by Barrett–Joyner–Halenda (BJH) method [Citation28]. Scanning electron microscopy (SEM) morphologies of samples were observed on a Philips XL-30ESEM. Transmission electron microscopy (TEM) morphologies of samples were observed with a Philips TEMCNAl-12 with an acceleration voltage of 100–120 kV.
3 Results and discussion
3.1 Synthesis condition
3.1.1 Effect of different crystallization temperatures on the phase of the composite
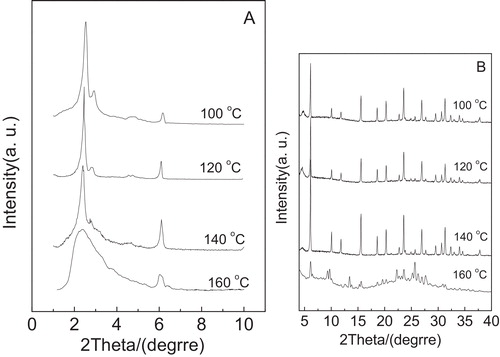
A presents the low angle XRD patterns of the calcined composite samples obtained after crystallization at 100, 120, 140 and 160 °C for 36 h with the addition of fluoride ions, respectively. As shown in A, the crystallization temperature is set at 100 °C, the four diffraction peaks, which can be assigned to (2 1 1), (2 2 0), (4 2 0) and (3 3 2) of the Ia3d cubic structure of MCM-48, are clearly noted. As the crystallization temperature increases to 120 °C, the four diffraction peaks appeared at ca. 2.46°, 2.82°, 4.54° and 4.74° are well resolved as compared with crystallization temperature of 100 °C, and the intensity becomes strong, suggesting that with aid of fluoride ions, the composite obtained synthesized at 120 °C for 36 h has the good mesoporous ordering. Different from the case at 120 °C, the crystallization temperature increased to 140 °C, the main diffraction peak (2 1 1) can be clearly observed and the shoulder peak (2 2 0) obviously weakened, indicating that the mesoporous ordering of the resulting composite decreased. However, the obtained composite sample did not have the cubic mesoporous framework of MCM-48 and finally changed into an amorphous phase when the crystallization temperature reached to 160 °C. The phenomenon may be due to the damage in self-assembly interaction between cation CTA+ and anion silicate aroused by higher crystallization temperature, which is unfavorable for the formation of the cubic framework of MCM-48 [Citation29]. The wide angle XRD patterns of the composite synthesized after crystallization at different temperatures for 36 h are shown in B. According to B, the crystallization temperature increased from 100 to 140 °C, the microporous characteristic peaks have almost no change. This indicates that the microporous phase in the composite has little effect at the temperature range. Furthermore, when the crystallization temperature increased to 160 °C, the crystallinity of the microporous phase in composite is rather low. Combined with A and B, the optimum crystallization temperature should be 120 °C.
Fig. 1 Low-angle XRD patterns (A) and high-angle XRD patterns (B) of the samples obtained after crystallization at different temperatures for 36 h.
3.1.2 Effect of different crystallization times on the phase of the composite
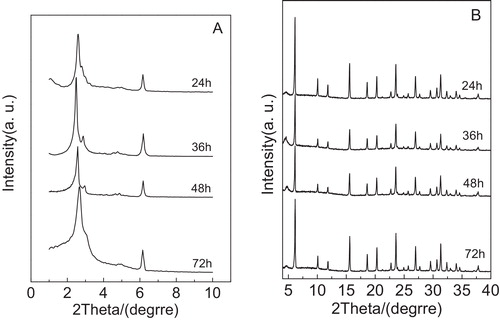
shows XRD patterns of the calcined samples synthesized after crystallization at 120 °C for different crystallization times (24, 36, 48 and 72 h). According to A, when the sample was crystallized at 120 °C for 24 h, the main diffraction peak (2 1 1) and the shoulder peak (2 2 0) were noted, which corresponded to the Ia3d cubic structure, but the characteristic peak (2 2 0) was very weak and several reflections of higher order for the sample cannot be observed, indicating that although the cubic framework in the composite is ultimately formed, the mesoporous ordering is poor. Different from the above case, the crystallization time increased to 36 h, the four diffraction peaks (2 1 1), (2 2 0), (4 2 0) and (3 3 2) were observed clearly and the diffraction peaks (2 1 1) and (2 2 0) became sharp and narrow and the intensities were strong, suggesting that the composite has good mesoporous ordering. When the crystallization time was 48 h, we found that the characteristic peaks (2 1 1), (2 2 0), (4 2 0) and (3 3 2) obviously weakened. However, the crystallization time is set at 72 h, the main peak (2 1 1) became wide and the shoulder peak (2 2 0) was very weak. At the same time, the diffraction peaks (4 2 0) and (3 3 2) appeared, indicating that the ordering of the MCM-48 mesoporous phase present in the composite deteriorated greatly. According to the Ref. [Citation15], Wang et al. reported that in the synthesis procedure of the pure silica MCM-48, the cubic phase can be ultimately formed after reaction for 72 h at 100 °C without fluoride ions addition. In the present study, the cubic phase of MCM-48 in composite molecular sieve is formed after reaction for 36 h at 120 °C with the aid of fluoride ions and the resulting composite molecular sieve exhibits good mesoporous ordering (see A). From the above discussion, it is concluded that the addition of fluoride ions can improve the structure ordering of mesoporous phase in composite molecular sieve. This phenomenon can be explained as follows: fluoride anions may play an catalytic role favoring the polycondensation of the silicate species, leading to enhancement of the polymerization degree of silicates in the presence of fluoride ions. Under this case, the enhancement of polymerization degree of silicates causes a high charge density of silicate network, which improves the interactions between the cationic micelles and the anionic framework. Thus, the ordering of mesoporous phase in composite molecular sieve can be enhanced [Citation30,Citation31]. Moreover, from B, it can be noted that the microporous characteristic peaks in composite have no effect in crystallization time range of 24–72 h.
Fig. 2 Low-angle XRD patterns (A) and high-angle XRD patterns (B) of the samples obtained after crystallization at 120 °C for different times.
3.2 XRD analysis of stability test
3.2.1 Hydrothermal stability
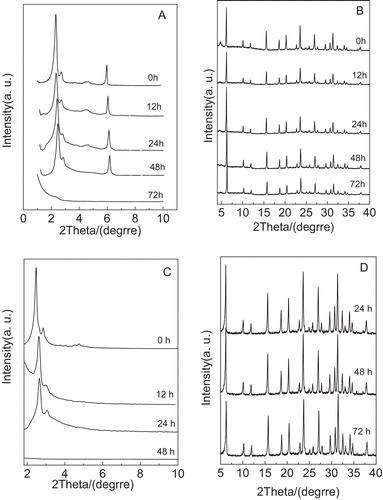
A illustrates the low-angle XRD patterns of the Y/MCM-48 sample after hydrothermal treatment at 100 °C for different times. As shown in A, it is clear that several characteristic peaks with the long range ordering appearing at 2θ = 2–6° can be found for the Y/MCM-48-24 sample, indicating that the cubic Ia3d framework in the Y/MCM-48-24 sample is still retained and has good mesoporous ordering. After hydrothermal treatment at 100 °C for 48 h, the Y/MCM-48-48 sample still possesses obvious characteristic peaks (2 1 1) and (2 2 0), but other low intense diffraction peaks of higher order cannot be seen easily, suggesting that the ordering of cubic mesoporous framework in the Y/MCM-48-48 sample decreased. However, when the hydrothermal treatment time increased to 72 h, the mesoporous characteristic peaks entirely disappeared, showing that the mesoporous framework in the Y/MCM-48 sample was collapsed completely. Also, with the prolonging in hydrothermal treatment time, the diffraction peaks shift to a higher 2θ value, indicating that the pore size of the resulting composite sample shrank, which agrees with the data listed in . The high-angle XRD patterns of the Y/MCM-48 sample after hydrothermal treatment at 100 °C for different times are shown in B. According to B, it is noted that several microporous characteristic peaks are still maintained in the XRD pattern of the Y/MCM-48-72 sample and the microporous phases present in several samples exhibit little change in the hydrothermal treatment time range of 12–72 h, certifying that the microporous phase in Y/MCM-48 composite still exhibits the better hydrothermal stability after hydrothermal treatment at 100 °C for 72 h. Further, the hydrothermal stability studies of the pure silica MCM-48 and NaY zeolite samples were also carried out and the results are presented in C and D, respectively. From C, we found that the mesoporous framework of the pure MCM-48 sample was entirely damaged after hydrothermal treatment at 100 °C for 48 h. Compared with the pure MCM-48 sample, the hydrothermal stability of the cubic mesoporous phase in Y/MCM-48 composite was improved, which is probably attributed to the presence of microporous phase in Y/MCM-48 composite. Moreover, it is observed from D that the characteristic peaks of the NaY sample are still retained after hydrothermal treatment at 100 °C for 72 h and the intensities of the diffraction peaks have almost no change, which is in agreement with the analysis results of the Y/MCM-48 composite.
Table 1 Surface areas, average pore sizes, total pore volumes and XRD analysis of various samples.
Fig. 3 XRD patterns of samples obtained after hydrothermal treatment at 100 °C for different times. (A) Low-angle XRD patterns of Y/MCM-48; (B) high-angle XRD patterns of Y/MCM-48; (C) low-angle XRD patterns of MCM-48; (D) high-angle XRD patterns of NaY.
3.2.2 Thermal stability
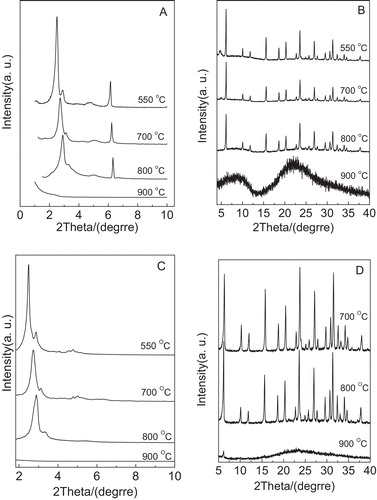
presents XRD patterns of several samples including Y/MCM-48, MCM-48 and NaY after thermal treatment at 700, 800 and 900 °C for 4 h, respectively. As displayed in A, the diffraction peaks (2 1 1) and (2 2 0) can be clearly noted even after the Y/MCM-48 sample was calcined at 800 °C for 4 h, suggesting that the Y/MCM-48 sample still has the cubic mesoporous framework of MCM-48. Compared with the Y/MCM-48 sample (after calcination at 550 °C), the ordering of the mesoporous phase in Y/MCM-48-800 sample decreased. However, when calcination temperature increased to 900 °C, it can be seen that the mesoporous diffraction peaks disappeared, showing that the cubic mesoporous structure in the Y/MCM-48 composite collapsed entirely. On the other hand, from B, we found that the microporous characteristic peaks are still retained even after calcination at 800 °C for 4 h. Different from the case at 800 °C, the microporous characteristic peaks disappeared after calcination at 900 °C for 4 h. Combined with A and B, it is concluded that after calcination at 800 °C for 4 h, the meso- and microporous structures of obtained sample are still maintained, suggesting that the Y/MCM-48 composite has good thermal stability. In addition, as shown in C, we found that the pure MCM-48 sample after calcination at 800 °C for 4 h still possesses obvious characteristic peaks (2 1 1) and (2 2 0); however, other low intense diffraction peaks (4 2 0 and 3 3 2) of higher order disappeared, indicating that the ordering of MCM-48 sample greatly decreased. After calcination at 900 °C for 4 h, the mesoporous framework of the MCM-48 sample collapsed completely. Further, according to D, it can be noticed that the NaY sample exhibits good thermal stability after calcination at 800 °C for 4 h.
Fig. 4 XRD patterns of the samples obtained after thermal treatment at different temperatures for 4 h. (A) Low-angle XRD patterns of Y/MCM-48; (B) high-angle XRD patterns of Y/MCM-48; (C) low-angle XRD patterns of MCM-48; (D) high-angle XRD patterns of NaY.
3.2.3 Acidic stability
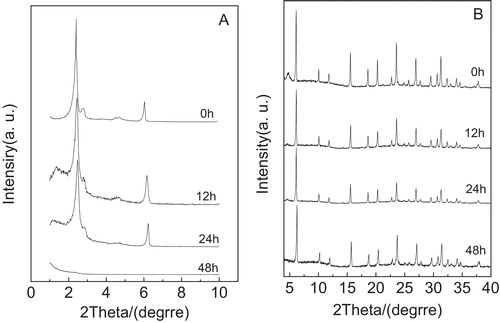
shows the low-angle XRD patterns (A) and high-angle XRD patterns (B) of the Y/MCM-48 sample after using 1 mol/L HCl solution treatment for 12, 24 and 48 h, respectively. As shown in A and B, we found that both the mesoporous and microporous characteristic peaks are still retained even after the Y/MCM-48 composite sample was treated in 1 mol/L HCl solution for 48 h and the ordering of mesoporous phase in the Y/MCM-48 composite was good. This indicates that the Y/MCM-48 sample possesses good stability in acidic solution. This may be due to the following factor: most of water molecules in solution combined with hydrogen ion to form hydroxonium ion, which decreased greatly the damage to Si–O–Si framework of the composite.
Fig. 5 Low-angle XRD patterns (A) and high-angle XRD patterns (B) of the samples obtained after treatment in 1 mol/L HCl solution for different times.
3.2.4 Basic stability
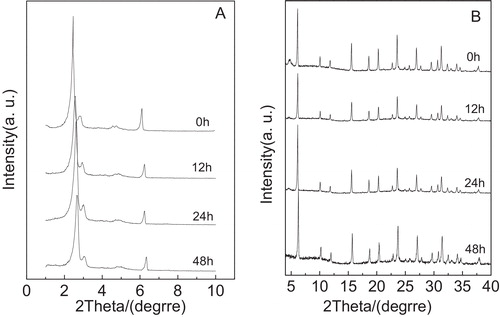
The XRD patterns of the Y/MCM-48 sample after treatment in 1 mol/L NaOH solution for different times are shown in . As displayed in A, it can be observed that after the Y/MCM-48 sample was treated in 1 mol/L NaOH solution for 24 h, the intensities of the diffraction peak (2 1 1) and the shoulder peak (2 2 0) slightly deteriorated and other low intense reflections of higher order appearing at 2θ = 4–6° cannot be clearly observed, suggesting that the ordering of mesoporous phase in the Y/MCM-48 composite decreased. However, the mesoporous characteristic peaks disappeared completely when the Y/MCM-48 sample was treated in 1 mol/L NaOH solution for 48 h. This indicates that the mesoporous framework in the Y/MCM-48 composite entirely collapsed at the case. On the other hand, from B, as compared with the parent composite, we found that the microporous characteristic peaks have no obvious change even after the Y/MCM-48 sample was treated in 1 mol/L NaOH solution for 48 h, suggesting that the microporous phase in composite exhibits good basic stability. Combined with A and A, after being treated for 48 h, the basic stability of the mesoporous phase in the Y/MCM-48 composite is poor as compared with the acidic stability. This may be attributed to the difference of hydrolysis manner of silicon species in mesoporous framework in acidic and basic solution.
Fig. 6 Low-angle XRD patterns (A) and high-angle XRD patterns (B) of the samples obtained after treatment in 1 mol/L NaOH solution for different times.
3.3 Nitrogen adsorption
3.3.1 Results of N2 adsorption after hydrothermal treatment
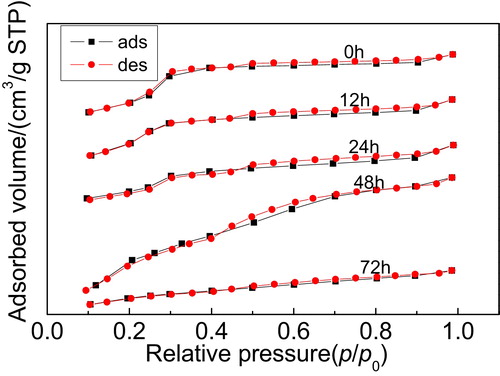
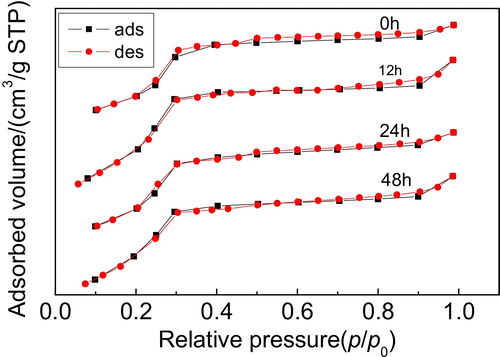
shows the N2 adsorption–desorption isotherms of the Y/MCM-48 sample after hydrothermal treatment at 100 °C for different times. As shown in , after hydrothermal treatment at 100 °C for 24 h, the isotherms of the obtained sample are typical type IV isotherms with hysteresis loop caused by capillary condensation in mesopores, indicating that the sample still possesses the mesoporous framework [Citation6]. After the Y/MCM-48 sample was hydrothermally treated at 100 °C for 48 h, the N2 adsorption–desorption isotherms did not belong to typical type IV isotherms, showing that the mesoporous structure of the sample was damaged partially. When the hydrothermal treatment time prolonged to 72 h, the isotherms of the obtained sample were not type IV isotherms; this is probably attributed to the damage of the mesoporous framework in the composite caused by longer hydrothermal treatment time.
Fig. 7 N2 adsorption–desorption isotherms of the samples obtained after hydrothermal treatment at 100 °C for different times.
3.3.2 Results of N2 adsorption after thermal treatment
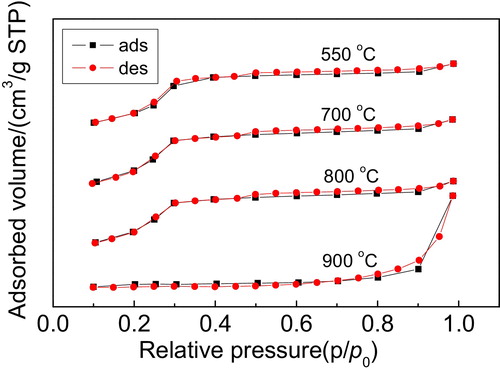
The N2 adsorption–desorption isotherms of the Y/MCM-48 sample after thermal treatment at different temperatures for 4 h are illustrated in . As shown in , it is noted that the isotherms of the sample obtained after thermal treatment at 800 °C for 4 h still exhibit the typical type IV isotherms, showing that the sample has obvious mesoporous structure. However, when the thermal treatment temperature was set at 900 °C, the isotherms of the Y/MCM-48-900 sample did not belong to type IV isotherms, combined with the BET surface area data (6 m2/g) listed in , suggesting that the mesoporous framework in the composite was entirely collapsed, which agrees with the results of XRD analysis.
Fig. 8 N2 adsorption–desorption isotherms of the samples obtained after thermal treatment at different temperatures for 4 h.
3.3.3 Results of N2 adsorption after acid treatment
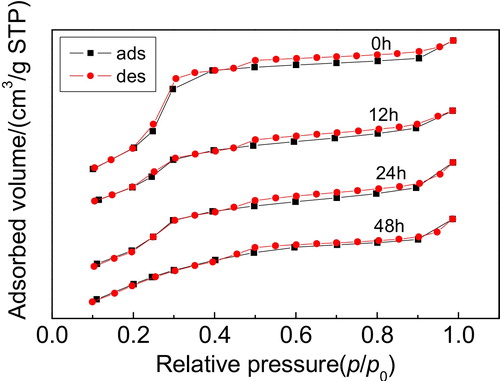
shows the N2 adsorption–desorption isotherms of the Y/MCM-48 sample after using 1 mol/L HCl solution treatment for different times. Clearly, it can be observed that after acid treatment for 48 h, the isotherms of the obtained sample exhibit typical type IV isotherms, showing that the sample still possesses the obvious mesoporous structure and further indicating that the Y/MCM-48 composite has good acidic stability.
Fig. 9 N2 adsorption–desorption isotherms of the samples obtained after treatment in 1 mol/L HCl solution for different times.
3.3.4 Results of N2 adsorption after base treatment
The N2 adsorption–desorption isotherms of the Y/MCM-48 sample after using 1 mol/L NaOH solution treatment for different times are illustrated in . From , the isotherms of the obtained sample after treatment in basic solution for 24 h are similar to the one of the parent Y/MCM-48 sample and belong to the typical type IV isotherms, certifying that the sample still retained good mesoporous structure. While the isotherms of the sample obtained after base treatment for 48 h are not typical type IV isotherms, suggesting that the mesoporous structure of the sample was damaged partially, we found that the mesoporous structure of the sample gradually lost with increase in base treatment time, and showing that the basic stability of the sample gradually decreased.
Fig. 10 N2 adsorption–desorption isotherms of the samples obtained after treatment in 1 mol/L NaOH solution for different times.
3.4 Textural properties of the samples
Textural properties of the samples are listed in . The mesoporous walls of samples were calculated from (a0/3.0919) − (pore size/2). From , compared with the MCM-48 sample, the surface area and pore volume of the Y/MCM-48 sample decreased. At the same time, we found that with increase in the calcination temperature, the prolonging in hydrothermal treatment, acid treatment and base treatment times, the surface area and pore volume of the obtained composite gradually decreased. Moreover, it can be noted that the mesoporous wall (6.85 nm) of the Y/MCM-48 sample is thicker than that of the MCM-48 sample (6.20 nm). This may be due to the wall of the composite containing the elementary and secondary building units of the Y type molecular sieve, indicating that the presence of the elementary and secondary building units in the wall of the composite is in favor of the improvement of stability of the mesoporous phase.
3.5 Morphology analysis of samples
Scanning electron microscopy (SEM) is used to determine the particle shape, particle morphology and particle size distribution of samples. presents the SEM images of NaY and Y/MCM-48 samples. From SEM image of the NaY sample (A), the octahedron crystal with particle size range of ca. 0.4–1 μm can be noted. Further, as shown in B, the morphological appearance of the Y/MCm-48 sample has a little similar with NaY sample.
Fig. 11 SEM images of the samples: (A) NaY zeolite and (B) Y/MCM-48.

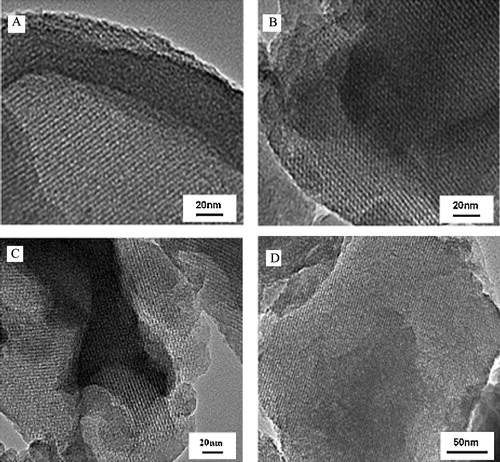
The TEM images of the several samples including the Y/MCM-48-48, Y/MCM-48-800, Y/MCM-48-48(A) and Y/MCM-48-48(B) are shown in . As shown in A, the obtained sample after hydrothermal treatment at 100 °C for 48 h still exhibits ordered arrays of mesopore with uniform pore size. Also, the sample obtained after calcination at 800 °C for 4 h has clearly the cubic mesoporous structure (B), further showing that the Y/MCM-48 composite has good thermal and hydrothermal stabilities. On the other hand, from C, after using 1 mol/L HCl treatment for 48 h, the composite sample still has obvious cubic mesoporous framework and good mesoporous ordering, suggesting that the Y/MCM-48 composite has good acidic stability. Although the sample obtained after using 1 mol/L NaOH treatment for 48 h maintained the mesoporous structure, the mesoporous ordering decreased (D). The results further indicate that the acidic stability of the Y/MCM-48 composite is superior to the basic stability.
Fig. 12 TEM images of the samples. (A) TEM image of the sample after hydrothermal treatment for 48 h; (B) TEM image of the sample after thermal treatment at 800 °C for 4 h; (C) TEM image of the sample after treatment in 1 mol/L HCl solution for 48 h; (D) TEM image of the sample after treatment in 1 mol/L NaOH solution for 48 h.
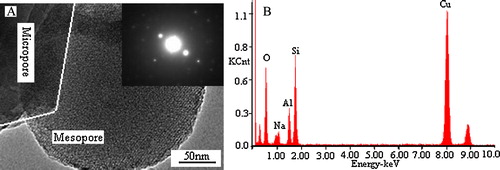
A presents the TEM image of the Y/MCM-48 sample. The inset in A is the SAED image of microphase in Y/MCM-48 sample. It is noticed from the TEM image that the Y/MCM-48 sample has both micropores and mesopores, and the inset SAED image proved that the core was typical and well structured NaY zeolite. Energy dispersive X-ray spectroscopy (EDS) was used to determine the chemical composition of microporous phase in Y/MCM-48 composite and the results are shown in B. As displayed in B, O, Na, Al, Si and Cu elements were detected, of which, Cu element was from the Cu net used in experiment.
Fig. 13 TEM image of the Y/MCM-48 sample (A) and EDS spectrum of the microphase in the Y/MCM-48 sample (B). The inset in (A) is the SAED image of microphase.
4 Conclusions
Y/MCM-48 composite molecular sieve with mesoporous and microporous structure was successfully synthesized by hydrothermal method with the aid of fluoride ions. The addition of fluoride ions can improve the ordering of the mesoporous phase in the composite. After the Y/MCM-48 composite was thermally treated at 800 °C for 4 h or hydrothermally treated at 100 °C for 48 h, the mesoporous structure in the composite is still maintained and the Y/MCM-48 composite has good thermal and hydrothermal stabilities. The acidic stability of the composite is better than the basic stability. Even after being treated in 1 mol/L HCl solution for 48 h, the obtained composite sample still has obvious mesoporous structure and good mesoporous ordering.
Notes
Peer review under responsibility of The Ceramic Society of Japan and the Korean Ceramic Society.
Related Research Data
References
- W.F.HöelderichG.HeitmannCatal. Today381997227233
- P.ProkešováS.MintovaJ.ČejkaT.BinMicroporous Mesoporous Mater.642003165174
- W.P.GuoL.M.HuangP.DengZ.Y.XueQ.Z.LiMicroporous Mesoporous Mater.44–452001427434
- M.J.VerhoefP.J.KooymanJ.C.Van der WaalM.S.RiguttoJ.A.PetersH.Van BekkumChem. Mater.132001683687
- Q.ZhaoQ.WangD.L.WuX.Q.FuT.S.JiangH.B.YinJ. Ind. Eng. Chem.18201213261331
- C.T.KresgeM.E.LeonowiczW.J.RothJ.C.VartuliJ.S.BeckNature3591992710712
- N.AoyamaT.YoshiharaS.I.FurukawaT.NittaH.TakahashiM.NakanoFluid Phase Equilib.2572007212216
- S.VeluL.WangM.OkazakiK.SuzukiS.TomuraMicroporous Mesoporous Mater.542002113126
- T.S.JiangL.D.LuX.J.YangQ.ZhaoT.TaoH.B.YinK.M.ChenJ. Porous Mater.1520086773
- R.WojcieszakS.MonteverdiM.MercyI.NowakM.M.BettaharAppl. Catal. A2682004241253
- M.C.KerbyT.F.DegnanD.O.MarlerJ.S.BeckCatal. Today10420055563
- Y.C.ChienH.P.WangS.H.LiuT.L.HsiungH.S.TaiC.Y.PengJ. Hazard. Mater.1512008461464
- C.G.JinG.LiX.S.WangY.WangL.X.ZhaoD.W.SunMicroporous Mesoporous Mater.1112008236242
- H.J.ZhangY.D.LiPowder Technol.18320087378
- L.Z.WangY.F.ShaoJ.L.ZhangM.AnpoMicroporous Mesoporous Mater.1002007241249
- T.S.JiangL.W.QiM.R.JiH.H.DingY.H.LiZ.F.TaoQ.ZhaoAppl. Clay Sci.62–6320123240
- P.ProkešováN.ŽilkováS.MintovaT.BeinJ.ČejkaAppl. Catal. A28120058591
- H.Y.ChenX.Y.CaiH.X.XiY.QianMicroporous Mesoporous Mater.1182009396402
- H.Y.XuJ.Q.GuanS.J.WuQ.B.KanJ. Colloid Interface Sci.3292009346350
- Y.S.OoiR.ZakariaA.R.MohamedS.BhatiaAppl. Catal. A27420041523
- P.Prokesova-FojtikovaS.MintovaJ.CejkaN.ZikovaA.ZukalMicroporous Mesoporous Mater.1–32006154160
- A.KarlssonM.StockerR.SchmidtMicroporous Mesoporous Mater.271999181192
- S.HabibF.LaunayS.LaforgeJ.D.ComparotA.FaustY.MillotT.OnfroyV.MontouilloutP.MagnouxJ.PaillaudA.GédéonAppl. Catal. A34420086169
- C.M.ZhangQ.LiuZ.XuK.WanMicroporous Mesoporous Mater.622003157163
- S.WangT.DouY.P.LiY.ZhangX.F.LiZ.C.YanCatal. Commun.620058791
- T.S.JiangD.L.WuJ.N.SongX.P.ZhouQ.ZhaoM.R.JiH.B.YinPowder Technol.2072011422427
- Y.H.ZhangY.C.LiuY.X.LiAppl. Catal. A34520087379
- E.P.BarrettL.G.JoynerP.P.HalendaJ. Am. Chem. Soc.731951373380
- Q.ZhaoX.P.ZhouM.R.JiD.L.WuT.S.JiangH.B.YinColloids Surf. A3842011513518
- A.C.VoegtlinF.RuchJ.L.GuthJ.PatarinL.HuveMicroporous Mater.9199795105
- Y.F.ShaoL.Z.WangJ.L.ZhangM.AnpoJ. Phys. Chem. B10920052083520841