
Abstract
The objective of the study was to develop and validate an analytical method for estimating the stability of duloxetine hydrochloride. The drug was subjected to the stress conditions prescribed by the International Conference on Harmonization, including hydrolysis, oxidation, photolysis and dry heat. Five degradation products were formed, which were separated by high-performance liquid chromatography on a Kromasil C18 (150 mm × 4.6 mm, 5 μm) column in a gradient elution programme. The flow rate was 1 ml/min, and the detection wavelength was set to 225 nm. The retention time of the drug was 35.7 min, and analysis was completed within 40 min. The method was validated with respect to linearity, precision, accuracy, robustness and limits of detection and quantification as per the International Conference on Harmonization. The results were linear (r2 = 0.999) over the range 50–400 μg/ml and accurate over the range 99.41–102.98. The method was robust and rugged, as there was insignificant variation in the results of analysis with changes in flow rate and temperature separately.
1 Introduction
Duloxetine ((+)-(s)-N-methyl-3-(1-naphthyloxy)-3-(thiophen-2-yl)-propan-1-amine) is a selective serotonin and norepinephrine reuptake inhibitor used primarily in the treatment of major depressive disorders and stress urinary incontinence [Citation1]. Duloxetine is also used to treat pain and tingling in diabetic neuropathy. Duloxetine, also known as LY248686 [Citation2,Citation3], is a potent dual inhibitor of reuptake of serotonin (5-hydroxytryptamine) and norepinephrine; its effect depends on binding to human serum albumin. It is approved by the United States Food and Drug Administration for the treatment of diabetic neuropathic pain.
Table 4 Inter-day precision.
Table 5 Accuracy.
Duloxetine hydrochloride is a highly lipophilic compound with strong base properties and a pKa of 9.5. Its structure is shown in . Phenolic impurities in duloxetine hydrochloride samples have been identified by mass spectrometry, nuclear magnetic resonance spectroscopy and X-ray analysis [Citation4–Citation6].
Fig. 1 Structure of duloxetine hydrochloride.
The purity of a drug product is determined on the basis of the percentage of the labelled amount of active pharmaceutical ingredient found in it by a suitable analytical method, which provides evidence for specificity, linearity range, accuracy, precision, detection limit, quantification limit, ruggedness and robustness of the method for regulatory purposes. High-performance liquid chromatography (HPLC) is the established technique for separating non-volatile organic compounds, drugs, metabolites and toxic residues by isocratic and gradient elution. Reverse-phase, ion-pair, ion and ion exchange HPLC and sometimes size exclusion chromatography are used conventionally. If the mobile phase remains constant throughout HPLC separation, the separation is deemed to be isocratic, while if the sample contains components of a wide range of polarities, gradient elution is the only way to elute all the compounds in the sample in a reasonable time while maintaining peak resolution by changing the ratio of polar to non-polar compounds in the mobile phase during the sample run. For a reverse-phase gradient, the solvent is initially relatively polar and slowly becomes more non-polar. A sample containing compounds with a wide range of polarities can be separated by gradient elution in a shorter time without loss of resolution in the earlier peaks or excessive broadening of later peaks; it is more difficult to maintain a constant flow rate with continuous changes in mobile phase composition. Gradient elution is used in preparative and large-scale chromatography for separation.
The parent drug stability test guideline Q1A (R2) issued by the International Conference on Harmonization (ICH) suggests that stress studies be carried out on a drug to establish its inherent stability [Citation7], i.e. the extent to which a product retains the same properties and characteristics it had at the time of packaging, within the specified limits, throughout storage and use [Citation8,Citation9]. Stability testing thus indicates the effect of environmental factors on the quality of a drug or a formulated product and is used to predict its shelf life, determine the proper storage conditions and suggest labelling instructions. Various analytical methods have been reported namely, HPLC [Citation10–Citation17], high-performance thin-layer chromatography [Citation18,Citation19], ultra-performance liquid chromatography [Citation20] and liquid chromatography–tandem mass spectrometry [Citation21–Citation23]. Degradation studies have also been reported [Citation24–Citation27] under thermal, acidic, alkaline, neutral hydrolysis and oxidative photolytic stress conditions.
2 Experimental
2.1 Materials
Pure duloxetine hydrochloride was donated by Unimark Remedies Ltd., Mumbai, India. Acetonitrile (HPLC grade) was purchased from Thomas Baker, Mumbai. Other chemicals used were of analytical grade. Ultrapure (double-distilled) water was obtained from a water purification unit.
2.2 Instrumentation
The HPLC instrument used was a Shimadzu LC-20 Prominence (Shimadzu, Kyoto, Japan) system equipped with an LC-20AD binary pump, a SPD-M20A photodiode array detector and a rheodyne injector. The output signal was monitored and processed with liquid chromatography solution software (Shimadzu, Kyoto, Japan). The injection volume was 20 μl, and chromatographic separation was achieved on a Kromasil C18 (150 mm × 4.6 mm), 5 μm particle size column.
2.3 Preparation of standard stock solution
A standard stock solution of the drug was prepared by dissolving pure drug in the mobile phase, i.e. 100 mg duloxetine hydrochloride in 100 ml methanol. The solution was sonicated and filtered through Whatman filter paper, and the resulting solution was further diluted with the mobile phase to a concentration of 1000 μg/ml.
2.4 Preparation of mobile phase
Solvent A: Potassium dihydrogen phosphate (0.68 g) was dissolved in 500 ml water, 0.1% triethylamine was added, and the pH was adjusted to 3.5 with orthophosphoric acid. Triethylamine was used to reduce tailing of the analyte. Methanol was added in proportions of 8.5:1.5. The solution was filtered through a 0.45-μm nylon 66 membrane filter and was further degassed in a sonicator for about 15 min. Solvent B was Methanol.
2.5 Degradation studies
All stress decomposition studies were performed at an initial drug concentration of 1000 μg/ml in methanol. Acid hydrolysis was performed in 0.5 N HCl at 80 °C for 9 h. The study under alkaline conditions was carried out in 1 N NaOH at 80 °C for 4 h. For the study under neutral conditions, drug dissolved in water was heated at 80 °C for 4 h. Oxidative stress studies were carried out at room temperature with 10%, 15% and 30% hydrogen peroxide for 24 h. For the photo-degradation studies, the solutions were exposed for 1 h in a chamber with 200–400 nm ultra-violet light and visible spectra (60,000–70,000 lx) for 10 h. Suitable controls were kept in the dark. Samples were withdrawn at appropriate times and analysed by HPLC after suitable dilution [Citation7,Citation24–Citation27]. Peak purity and mass balance were monitored for each degradation condition.
2.6 Separation studies
Before method development, various physicochemical parameters must be known, such as the pKa, log P, solubility, absorptivity and wavelength maximum of the drug. The pKa is important as most pH-related changes in retention occur at pH values within 1.5 units of the pKa value [Citation28]. Studies were first conducted on all reaction solutions individually and then on a mixture of the solutions, in which decomposition was observed. Separation was achieved by gradient elution. Initial studies on individual reaction solutions were conducted with methanol:potassium dihydrogen phosphate buffer and triethylamine (0.1%) (pH adjusted to 3.5 with orthophosphoric acid), with the gradient programme shown in .
Table 1 Gradient programme for separation studies.
2.7 Validation of the method [Citation29,Citation30]
2.7.1 Linearity and range
The stock solution was diluted with mobile phase to prepare solutions containing 50–400 μg/ml of the drug. Each concentration was injected six times onto the HPLC column, and the peak areas were noted and plotted against the corresponding concentration to obtain a calibration curve.
2.7.2 Precision
Injections of three concentrations (100, 200 and 300 μg/ml), repeated six times, were given on the same day, and the relative standard deviation was calculated to determine intra-day precision.
2.7.3 Accuracy
Recovery studies by the standard addition method were performed to determine the accuracy of the method, with three concentrations (50%, 100% and 150%) of spiked drug.
2.7.4 Specificity and selectivity
The specificity of the method for the drug was established by determining interference (if any) from solvent or degradation products, with resolution of the drug peak from the nearest resolving peaks. Overall selectivity was established by determining the purity of the peak.
2.7.5 Robustness
The effects of varying the mobile phase composition, flow rate and wavelength were studied.
2.7.6 Limits of detection and quantification
The limit of detection (LOD) is defined as the lowest concentration of an analyte that can be detected in a sample, whereas the limit of quantification (LOQ) is the lowest concentration of an analyte that can be determined with acceptable precision and accuracy in a sample under the stated operational conditions. LOD and LOQ were calculated according to the formulae given in the ICH guidelines.
3 Results and discussion
HPLC studies on duloxetine hydrochloride under different stress conditions suggested the following degradation behaviour. The drug was found to be susceptible to acid hydrolysis (0.5 N HCl, 9 h), with 5–16% degradation when heated at 80 °C, but was stable in neutral hydrolysis (water, 4 h). It was stable to degradation with 10%, 15% and 30% hydrogen peroxide at room temperature for 24 h.
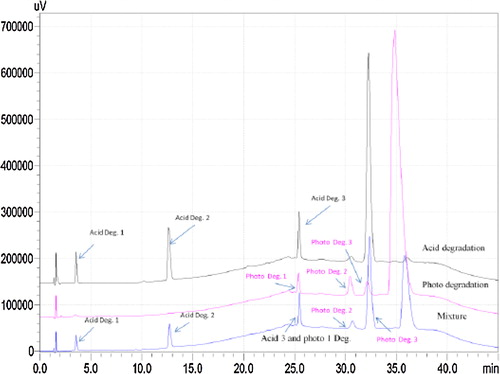
Three major degradation products were observed after exposure of drug solution to ultra-violet light for 33 min, with five major degradation peaks at retention times of 3.5 min (Degr. 1), 12.7 min (Degr. 2), 25.4 min (Degr. 3), 30.6 min (Degr. 4) and 32.4 min (Degr. 5) (). The spectral data showed that acid Degr. 3 and photolytic Degr. 1 were identical ().
Fig. 2 Chromatogram of degradation products and active pharmaceutical ingredient of duloxetine.
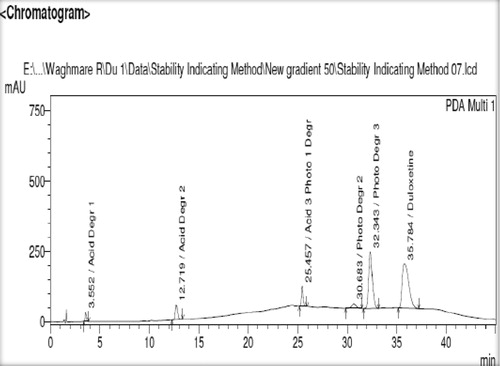
Fig. 3 Ultra-violet spectra of acid Degr. 3 and photolytic Degr. 1.

The solid-state studies showed that duloxetine hydrochloride is stable to the effect of temperature. When the drug powder was exposed to dry heat at 80 °C for 8 days, no decomposition was seen.
The method was optimised to separate the major degradation products formed under various conditions, and resolution was checked with a mixture of the degradation solutions to confirm the separation. The resulting chromatograms are shown in and . They indicate that the method successfully separated the drug from all degradation products.
Fig. 4 Chromatogram of acid and photolytic degradation products.
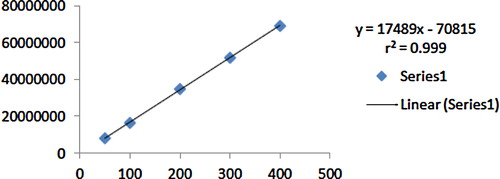
The method was validated as per the ICH guidelines. The linearity of the peak area response was determined by injecting samples of 20 μl of dilutions of the standard stock solution onto the column at a flow rate 1.0 ml/min. Each dilution was injected six times. Drug in the eluates was monitored at 225 nm, and the corresponding chromatograms were obtained. The regression of the plot was obtained by least squares regression. The mean peak areas were calculated from these chromatograms, and a plot of concentration over the peak area was obtained ().
Table 2 Linearity of peak areas of duloxetine hydrochloride.
The peak area was plotted against the corresponding concentrations to obtain the calibration curve ().
Fig. 5 Calibration curve of duloxetine hydrochloride on HPLC.
As the relative SD for inter-day precision was less than 2, it can be concluded that the method is reproducible ().
Table 3 Intra-day precision.
Accuracy tested by the standard addition method was in the range of 99.41–102.48% ( and ).
As small deliberate changes did not result in significant changes in the peak area, the method is robust and resistant to such changes ().
Table 6 Robustness.
The LOD and LOQ were 1.17 and 3.57 μg/ml, respectively, as confirmed by preparing concentrations below and above these values.
4 Conclusion
The HPLC method reported here is accurate, precise, reproducible, specific and stable. There is allowable variation in flow rate, temperature, pH and mobile phase composition, which indicate that the method is robust. The low relative SD for the percentage assay of the test preparation shows that the proposed method is rugged.
This study shows that the drug is highly sensitive to degradation with acid and photolytic stress but stable in dry heat and oxidative conditions. This method allows separation of all degradation products formed under a variety of conditions.
Acknowledgements
The authors are thankful to Unimark Remedies Ltd, Mumbai, India, for providing a sample of duloxetine hydrochloride. The authors are also thankful to the Principal, Sinhgad Institute of Pharmacy, Narhe, Pune 411041, for providing the facilities required for completion of this research.
Notes
Peer review under responsibility of Taibah University.
Related Research Data
References
- M.J.SkibicL.A.KingM.KhanP.J.FoxB.E.WingerS.W.BaertschiArtifactual formylation of the secondary amine of duloxetine hydrochloride by acetonitrile in the presence of titanium dioxide: implications for HPLC method developmentJ. Pharm. Biomed. Anal.532010432439
- N.V.V.S.S.RamanK.A.HarikrishnaA.V.S.S.PrasadaR.ReddyK.RamakrishnaDetermination of duloxetine hydrochloride in the presence of process and degradation impurities by a validated stability-indicating RP-LC methodJ. Pharm. Biomed. Anal.512010994997
- L.XiangpingD.YingxiangStudy on the binding of chiral drug duloxetine hydrochloride to human serum albuminEur. J. Med. Chem.45201040434049
- E.BrennaS.FrigoliG.FronzaC.FugantiL.MalpezziIsolation and characterization of a phenolic impurity in a commercial sample of duloxetineJ. Pharm. Biomed. Anal.43200715731575
- M.YunoosD.G.SankarB.P.KumarS.HameedA.HussainSimple UV spectrophotometric determination of duloxetine hydrochloride in bulk and in pharmaceutical formulationsE-J. Chem.72010785788
- R.V.A.RajT.RameshA.P.KumarA validated UV spectrophotometric determination of an antidepressant drug – duloxetine hydrochloride from capsule formulationsInt. J. Pharma Bio Sci.22011716720
- S.SinghB.SinghR.BahugunaL.WadhwaR.J.SaxenaStress degradation studies on ezetimibe and development of a validated stability-indicating HPLC assayJ. Pharm. Biomed. Anal.41200610371040
- S.BajajD.SinglaN.SakhujaStability testing of pharmaceutical productsJ. Appl. Pharm. Sci.22012129138
- T.PranshuB.BhanuWHO role and guidelines in stability study of pharmaceuticals: a regulatory perspectiveInt. J. Res. Pharmaceut. Biomed. Sci.3201213791386
- J.T.JohnsonS.W.OldhamR.J.LantzA.F.DeLongHigh performance liquid chromatographic method for the determination of duloxetine and desmethyl duloxetine in human plasmaJ. Liq. Chromatogr. Relat. Technol.19199616311641
- B.A.OlsenM.D.ArgentineHPLC method development for duloxetine hydrochloride using a combination of computer-based solvent strength optimization and solvent selectivity mixture designJ. Liq. Chromatogr. Relat. Technol.19199619932007
- S.PankajT.T.MariappanU.C.BanerjeeHigh performance liquid chromatographic method for the simultaneous estimation of the key intermediates of duloxetineTalanta672005975978
- L.MercoliniR.MandrioliR.CazzollaM.AmoreM.A.RaggiHPLC analysis of the novel antidepressant duloxetine in human plasma after an original solid-phase extraction procedureJ. Chromatogr. B85620078187
- C.WaldschmittF.VogelC.MaurerC.HiemkeMeasurement of duloxetine in blood using high-performance liquid chromatography with spectrophotometric detection and column switchingTher. Drug Monit.292007767772
- S.DasariR.K.ViriyalaK.SantoshA.KumariB.V.V.RavikumarS.P.S.BishtA validated RP-HPLC method for the analysis of duloxetine hydrochloride in pharmaceutical dosage formsPharm. Glob. (IJCP)1201013
- P.S.LakshmanaM.SrinivasanS.ThiyagarajanQ.MarinaDetermination of duloxetine hydrochloride in pharmaceutical formulation by HPLC with UV detectionInt. J. Chem. Technol. Res.2201014411444
- P.P.DahivelkarV.K.RedasaniS.B.BariStudy of hydrolytic and oxidative behavior of duloxetine in aqueous solution by RP-HPLCJ. Global Pharma Technol.220101923
- S.S.DhaneshwarP.DeshpandeM.PatilG.VadnerkarS.R.DhaneshwarDevelopment and validation of HPTLC method for estimation of duloxetine hydrochloride in bulk drug tablet dosage formIndian J. Pharmaceut. Sci.702008233236
- S.SheikhA.W.SiddiquiM.T.MasroorV.AroraStability-Indicating HPTLCMethod for Determination of Duloxetine Hydrochloride in Bulk Drug and Tablet FormulationChromatogr. Res. Int.2011201115
- U.K.ChhalotiyaK.K.BhattD.A.ShahS.L.BaldaniaDevelopment and validation of a stability-indicating RP-HPLC method for duloxetine hydrochloride in its bulk and tablet dosage formSci. Pharm.782010857868
- N.MaB.K.ZhangH.D.LiB.M.ChenP.XuF.WangR.H.ZhuS.FengD.X.XiangY.G.ZhuDetermination of duloxetine in human plasma via LC/MS and subsequent application to a pharmacokinetic study in healthy Chinese volunteersClin. Chim. Acta3802007100105
- S.P.SenthamilK.V.GowdaU.MandalS.W.D.SamT.K.PalDetermination of duloxetine in human plasma by liquid chromatography with atmospheric pressure ionization–tandem mass spectrometry and its application to pharmacokinetic studyJ. Chromatogr. B: Analyt. Technol. Biomed. Life Sci.8582007269275
- D.C.ReddyA.T.BapujiV.S.RaoV.HimabinduR.D.RamaS.SyedbaH.L.V.RavikiranDevelopment and Validation of a LC/MS/MS Method for the Determination of Duloxetine in Human Plasma and its Application to Pharmacokinetic StudyE-J. Chem.92011899911
- V.R.SinhaA.R.KumriaJ.R.BhingeStress degradation studies on duloxetine hydrochloride and development of an RP-HPLC method for its determination in capsule formulationJ. Chromatogr. Sci.472009589593
- C.N.BhimanadhuniD.R.GarikapatiC.SrinivasDevelopment and validation of RP-HPLC method for determination of duloxetine hydrochloride in bulk and dosage formInt. Curr. Pharmaceut. J.1201298102
- S.ShahnawazA.SiddiquiM.T.MasroorV.AroraStability-indicating HPTLC method for determination of duloxetine hydrochloride in bulk drug and tablet formulationChromatogr. Res. Int.2011201115
- V.A.ReddyS.B.M.UlareddyK.C.RaoB.MadhusudhanreddyStability indicating nature of RP-HPLC method for determination of impurity profile and degradation impurities in duloxetine hydrochlorideDer Pharma Chem.4201217351741
- S.SinghM.BakshiDevelopment of validated stability-indicating assay methods-critical reviewJ. Pharm. Biomed. Anal.28200210111040
- International Conference on Harmonization Q1A (R2), Stability testing of new drug substances and products, Geneva, 2003.
- International Conference on Harmonization Q2 (R1), Validation of analytical procedures: text and methodology, Geneva, 2005.