
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
New functionalized N-heterocyclic derivatives were synthesized from an itaconic anhydride (ITA) precursor and characterized by spectral and analytical data. All of the synthesized products were screened for in vitro antioxidant and antimicrobial properties. Among the tested compounds, derivatives 5a and 8a exhibited noticeable DPPH radical scavenging activity, reducing power capacity and nitric oxide scavenging activity compared to standard glutathione. Compound 3d showed strong antibacterial activity, whereas compounds 3a and 4a exhibited potential antifungal activity. Compounds exhibiting good antioxidant activity were docked with tyrosinase enzyme (PDB ID: 3NM8) from Bacillus megaterium to predict their putative interactions.
1 Introduction
Antioxidants are molecules that are capable of inhibiting the oxidation of other molecules and preventing cell death after the release of free radicals. The search for new molecules with antioxidant properties is an exceptionally active area of research, as such molecules have the potential to reduce the risk of many chronic diseases, including atherosclerosis, stroke, diabetes, Alzheimer's disease, and some forms of cancer. Furthermore, oxidative stress is responsible for DNA, protein and membrane damage [Citation1]. Different types of antioxidants (vitamins C and E, glutathione, lipoic acid, butylated phenols, etc.) are widely used in the chemical and pharmaceutical industries to interrupt radical-chain oxidation processes. Hence, there is great scientific interest in the discovery of efficient synthetic and/or natural antioxidants [Citation2,Citation3].
Synthetic compounds, including imides [Citation4], pyridazines [Citation5], thiazoles [Citation6], and benzimidazoles [Citation7], have been exploited for their antioxidant and antimicrobial activities. Moreover, compounds with groups capable of providing free electrons (e.g., –S, –O, or –N), either in the form of a negative charge or a lone pair of electrons, may show higher antioxidant activity due to their redox properties [Citation8].
Based on the above observations and the need for ongoing efforts to synthesize a large range of biologically active heterocycles [Citation9], the objectives of the present study were to synthesize various heterocyclic derivatives, such as imides, pyridazines, thiazoles, and benzimidazoles, from a single precursor (itaconic anhydride (ITA)) and to evaluate the in vitro and in silico antioxidant and antimicrobial properties of the new heterocyclic derivatives.
2 Experiments
2.1 Materials and methods
The melting points were measured in open capillary tubes and were not corrected. The purity of the compounds was confirmed by thin-layer chromatography using Merck silica gel 60 F254-coated aluminium plates. The IR spectra were recorded on a Shimadzu-FTIR Infrared spectrometer in KBr (νmax in cm−1). The 1H NMR (400 MHz) spectra were recorded on a Varian 400 spectrometer with 5 mm PABBO BB-1H TUBES. The 13C NMR (100 MHz) spectra were recorded for approximately 0.03 M solutions in DMSO at 100 MHz with CDCl3 as the internal standard. LCMS was completed using the Agilent 1200 series LC and a Micromass zQ spectrometer. GCMS was completed using the Perkin Elmer (Clarus 680 model) GC and a Clarus 600 (EI) spectrometer. Elemental analysis was carried out with the VARIO EL-III (Elementar Analysensysteme, GmBH).
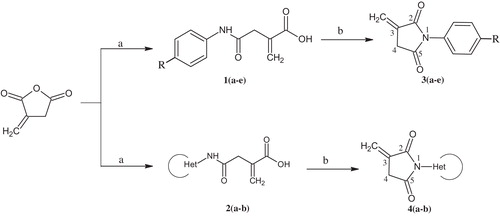
2.2 General procedure for the synthesis of substituted oxobutanoic acid derivatives 1(a-e), 2(a-b) ()
Itaconic anhydride (0.112 g, 1 mmol) was dissolved in acetone (30 mL). The substituted/heterocyclic amine (1 mmol) was added in small portions under stirring at room temperature over a time span of 30 min. The mixture became a yellow slurry. Stirring was continued for 1.5 h, after which the solution was filtered. The obtained solid was washed with acetone and dried. The crude products were further purified by a recrystallization method with methanol as a solvent.
Scheme 1 Synthetic pathways to substituted/heterocyclic imides. (a) Substituted/heterocyclic amines/MDC, stirring; (b) AcOH, reflux.
2.2.1 4-[(4-Chlorophenyl)amino]-2-methylidene-4-oxobutanoic acid (1a)
IR (KBr): νmax (cm−1), 3280 (amide NH), 3088 (carboxylic OH), 3020 (Ar–H), 2918 (aliphatic C–H), 1681 (carboxylic C=O), 1668 (amide C=O), 1631 (C=N), 1577 (C=C), 808 (C–Cl). 1H NMR (400 MHz, DMSO-d6): δ ppm, 2.73 (s, 2H, O=C–CH2), 5.74 (d, 1H, HA, JAB = 1.2 Hz), 6.17 (d, 1H, HB, JAB = 1.2 Hz), 7.40–7.64 (m, 4H, Ar–H), 10.13 (s, 1H, NH), 10.30 (s, 1H, OH). LCMS (m/z): 240 (M++1). Calcd. for C11H10ClNO3: C, 55.13; H, 4.21; N, 5.84; Found: C, 55.11%; H, 4.23%; N, 5.82%.
2.2.2 4-[(4-Fluorophenyl)amino]-2-methylidene-4-oxobutanoic acid (1b)
IR (KBr): νmax (cm−1), 3282 (amide NH), 3088 (carboxylic OH), 3021 (Ar–H), 2919 (aliphatic C–H), 1712 (carboxylic C=O), 1682 (amide C=O), 1633 (C=N), 1577 (C=C), 728 (C-F). 1H NMR (400 MHz, DMSO-d6): δ ppm, 2.73 (s, 2H, O=C–CH2), 5.74 (d, 1H, HA, JAB = 1.2 Hz), 6.17 (d, 1H, HB, JAB = 1.2 Hz), 7.40–7.64 (m, 4H, Ar–H), 10.10 (s, 1H, NH), 10.30 (s, 1H, OH). LCMS (m/z): 224 (M++1). Calcd. for C11H10FNO3: C, 59.19; H, 4.52; N, 6.28; Found: C, 59.17%; H, 4.54%; N, 6.26%.
2.2.3 4-[(4-Bromophenyl)amino]-2-methylidene-4-oxobutanoic acid (1c)
IR (KBr): νmax (cm−1), 3211 (amide NH), 3155 (carboxylic OH), 3024 (Ar–H), 2887 (aliphatic C–H), 1712 (carboxylic C=O), 1685 (amide C=O), 1639 (C=N), 1584 (C=C), 688 (C-Br). 1H NMR (400 MHz, DMSO-d6): δ ppm, 2.73 (s, 2H, O=C–CH2), 5.74 (d, 1H, HA, JAB = 1.2 Hz), 6.17 (d, 1H, HB, JAB = 1.2 Hz), 7.40–7.64 (m, 4H, Ar–H), 10.10 (s, 1H, NH), 10.30 (s, 1H, OH). LCMS (m/z): 285 (M++1), 286 (M++2). Calcd. for C11H10BrNO3: C, 46.50; H, 3.55; N, 4.93; Found: C, 46.48%; H, 3.57%; N, 4.91%.
2.2.4 4-[(4-Iodophenyl)amino]-2-methylidene-4-oxobutanoic acid (1d)
IR (KBr): νmax (cm−1), 3284 (amide NH), 3194 (carboxylic OH), 3101 (Ar–H), 2920 (aliphatic C–H), 1708 (carboxylic C=O), 1695 (amide C=O), 1633 (C=N), 1519 (C=C), 503 (C–I). 1H NMR (400 MHz, DMSO-d6): δ ppm, 2.73 (s, 2H, O=C–CH2), 5.74 (d, 1H, HA, JAB = 1.2 Hz), 6.17 (d, 1H, HB, JAB = 1.2 Hz), 7.41–7.64 (m, 4H, Ar–H), 10.13 (s, 1H, NH), 10.30 (s, 1H, OH). LCMS (m/z): 332 (M++1). Calcd. for C11H10INO3: C, 39.90; H, 3.04; N, 4.23; Found: C, 39.88%; H, 3.06%; N, 4.21%.
2.2.5 2-Methylidene-4-[(4-methylphenyl)amino]-4-oxobutanoic acid (1e)
IR (KBr): νmax (cm−1), 3285 (amide NH), 3184 (carboxylic OH), 3100 (Ar–H), 2923 (aliphatic C–H), 1718 (carboxylic C=O), 1685 (amide C=O), 1625 (C=N), 1510 (C=C). 1H NMR (400 MHz, DMSO-d6): δ ppm, 2.35 (s, 3H, CH3), 2.73 (s, 2H, O=C–CH2), 5.74 (d, 1H, HA, JAB = 1.2 Hz), 6.17 (d, 1H, HB, JAB = 1.2 Hz), 7.40–7.64 (m, 4H, Ar–H), 10.10 (s, 1H, NH), 10.30 (s, 1H, OH). LCMS (m/z): 219 (M+). Calcd. for C12H13NO3: C, 65.74; H, 5.98; N, 6.39; Found: C, 65.72%; H, 6.00%; N, 6.37%.
2.2.6 2-Methylidene-4-oxo-4-(1,3-thiazol-2-ylamino)butanoic acid (2a)
IR (KBr): νmax (cm−1), 3111 (amide NH), 3053 (carboxylic OH), 2989 (Ar–H), 2942 (aliphatic C–H), 1705 (carboxylic C=O), 1668(amide C=O), 1635 (C=N), 1581 (C=C). 1H NMR (400 MHz, DMSO-d6): δ ppm, 2.73 (s, 2H, O=C–CH2), 5.59 (d, 1H, HA, JAB = 1.6 Hz), 6.31 (d, 1H, HB, JAB = 1.6 Hz), 7.17–7.58 (m, 2H, Ar–H), 9.12 (s, 1H, NH), 9.68 (s, 1H, OH). LCMS (m/z): 215 (M++1). Calcd. for C8H10N2O3S: C, 44.85; H, 4.70; N, 13.08; Found: C, 44.83%; H, 4.72%; N, 13.06%.
2.2.7 4-(1,3-Benzothiazol-2-ylamino)-2-methylidene-4-oxobutanoic acid (2b)
IR (KBr): νmax (cm−1), 3294 (amide NH), 3116 (carboxylic OH), 3035 (Ar–H), 2920 (aliphatic C–H), 1683 (carboxylic C=O), 1654 (amide C=O), 1629 (C=N), 1525 (C=C). 1H NMR (400 MHz, DMSO-d6): δ ppm, 2.83 (s, 2H, O=C–CH2), 5.57 (d, 1H, HA, JAB = 1.6 Hz), 6.30 (d, 1H, HB, JAB = 1.6 Hz), 7.17–7.59 (m, 4H, Ar–H), 9.10 (s, 1H, NH), 9.68 (s, 1H, OH). LCMS (m/z): 263 (M++1). Calcd. for C10H10N2O3S: C, 54.95; H, 3.84; N, 10.68; Found: C, 54.93%; H, 3.86%; N, 10.66%.
2.3 General procedure for the synthesis of substituted imides 3(a-e), 4(a-b) ()
A mixture of anhydrous NaOAc (5 mmol) and acetic anhydride (20 mmol) was heated with stirring until all of the NaOAc was dissolved. Substituted oxobutanoic acid was added to the reaction mixture and stirred for 30–40 min. After completion of the reaction, the mixture was allowed to cool to room temperature and quenched in ice-cold water. The precipitate was filtered and dried, and the crude product was purified by column chromatography using DCM as the eluent ().
2.3.1 1-(4-Chlorophenyl)-3-methylidenepyrrolidine-2,5-dione (3a)
IR (KBr): νmax (cm−1), 3005 (Ar–H), 2964 (aliphatic C–H), 1716 (C=O), 1512 (C=C), 1394 (C=N), 829 (C–F). 1H NMR (400 MHz, DMSO-d6): δ ppm, 3.54 (s, 2H, CH2), 5.78 (d, 1H, HA, JAB = 2.0 Hz), 6.31 (d, 1H, HB, JAB = 2.0 Hz), 7.17–7.38 (m, 4H, Ar–H). 13C NMR (100 MHz, DMSO): δ ppm, 172.77 (pyrrolidine 5C=O), 169.42 (pyrrolidine 2C=O), 132.74 (C=CH2) 127.80, 125.44, 122.71, 115.97 (aromatic C's), 122.09 (methylidine CH2), 34.93 (pyrrolidine 4 C). TLC (6:4) hexane/EtOAc, Rf = 0.29; GCMS (m/z): 221 (M+), 222 (M++1). Calcd. for C11H8ClNO2: C, 59.61; H, 3.64; N, 6.32; Found: C, 59.59%; H, 3.66%; N, 6.30%.
2.3.2 1-(4-Fluorophenyl)-3-methylidenepyrrolidine-2,5-dione (3b)
IR (KBr): νmax (cm−1), 3010 (Ar–H), 2962 (aliphatic C–H), 1710 (C=O), 1522 (C=C), 1414 (C=N), 718 (C–F). 1H NMR (400 MHz, DMSO-d6): δ ppm, 3.55 (s, 2H, CH2), 5.78 (d, 1H, HA, JAB = 2.0 Hz), 6.31 (d, 1H, HB, JAB = 2.0 Hz), 7.17–7.38 (m, 4H, Ar–H). 13C NMR (100 MHz, DMSO): δ ppm, 172.77 (pyrrolidine 5C=O), 169.42 (pyrrolidine 2C=O), 161.00 (C–F), 132.74 (C=CH2) 127.80, 122.71, 115.97 (aromatic C's), 122.09 (methylidine CH2), 34.93 (pyrrolidine 4 C). TLC (6:4) hexane/EtOAc, Rf = 0.25; GCMS (m/z): 205 (M+), 206 (M++1). Calcd. for C11H8FNO2: C, 64.39; H, 3.93; N, 6.83; Found: C, 64.37%; H, 3.95%; N, 6.81%.
2.3.3 1-(4-Bromophenyl)-3-methylidenepyrrolidine-2,5-dione (3c)
IR (KBr): νmax (cm−1), 3005 (Ar–H), 2964 (aliphatic C–H), 1716 (C=O), 1512 (C=C), 1394 (C=N), 688 (C-Br). 1H NMR (400 MHz, DMSO-d6): δ ppm, 3.55 (s, 2H, CH2), 5.78 (d, 1H, HA, JAB = 2.0 Hz), 6.31 (d, 1H, HB, JAB = 2.0 Hz), 7.17–7.38 (m, 4H, Ar–H). 13C NMR (100 MHz, DMSO): δ ppm, 172.77 (pyrrolidine 5C=O), 169.42 (pyrrolidine 2C=O), 132.74 (C=CH2) 127.80, 125.44, 122.71, 115.97 (aromatic C's), 122.09 (methylidine CH2), 34.93 (pyrrolidine 4 C). TLC (6:4) hexane/EtOAc, Rf = 0.31; GCMS (m/z): 266 (M+), 267 (M++1). Calcd. for C11H8BrNO2: C, 49.65; H, 3.03; N, 5.26; Found: C, 49.63%; H, 3.05%; N, 5.24%.
2.3.4 1-(4-Iodophenyl)-3-methylidenepyrrolidine-2,5-dione (3d)
IR (KBr): νmax (cm−1), 3012 (Ar–H), 2964 (aliphatic C–H), 1716 (C=O), 1512 (C=C), 1394 (C=N), 572 (C–I). 1H NMR (400 MHz, DMSO-d6): δ ppm, 3.54 (s, 2H, CH2), 5.78 (d, 1H, HA, JAB = 2.0 Hz), 6.31 (d, HB, JAB = 2.0 Hz), 7.17–7.38 (m, 4H, Ar–H). 13C NMR (100 MHz, DMSO): δ ppm, 172.77 (pyrrolidine 5C=O), 169.42 (pyrrolidine 2C=O), 132.74 (C=CH2) 127.80, 122.71, 115.97, 108.67 (aromatic C's), 122.09 (methylidine CH2), 34.93 (pyrrolidine 4 C). TLC (6:4) hexane/EtOAc, Rf = 0.34; GCMS (m/z): 313 (M+), 314 (M++1). Calcd. for C11H8INO2: C, 42.20; H, 2.58; N, 4.47; Found: C, 42.18%; H, 2.60%; N, 4.45%.
2.3.5 3-Methylidene-1-(4-methylphenyl)pyrrolidine-2,5-dione (3e)
IR (KBr): νmax (cm−1), 3010 (Ar–H), 2964 (aliphatic C–H), 1715 (C=O), 1526 (C=C), 1410 (C=N). 1H NMR (400 MHz, DMSO-d6): δ ppm, 2.45 (s, 3H, –CH3), 3.52 (s, 2H, CH2), 5.78 (d, 1H, HA, JAB = 2.0 Hz), 6.31 (d, 1H, HB, JAB = 2.0 Hz), 7.17–7.38 (m, 4H, Ar–H). 13C NMR (100 MHz, DMSO): δ ppm, 172.77 (pyrrolidine 5C=O), 169.42 (pyrrolidine 2C=O), 132.74 (C=CH2) 127.80, 125.44, 122.71, 115.97 (aromatic C's), 122.09 (methylidine CH2), 34.93 (pyrrolidine 4 C). 24.03 (CH3). TLC (6:4) hexane/EtOAc, Rf = 0.27; GCMS (m/z): 201 (M+). Calcd. for C12H11NO2: C, 71.63; H, 5.51; N, 6.96; Found: C, 71.61%; H, 5.53%; N, 6.94%.
2.3.6 3-Methylidene-1-(1,3-thiazol-2-yl)pyrrolidine-2,5-dione (4a)
IR (KBr): νmax (cm−1), 3057 (Ar–H), 2960 (aliphatic C–H) 1695 (C=O), 1544 (C=C), 1444 (C=N). 1H NMR (400 MHz, DMSO-d6): δ ppm, 2.40 (s, 2H, CH2), 5.59 (d, 1H, HA, JAB = 1.6 Hz), 6.31 (d, 1H, HB, JAB = 1.6 Hz), 7.17–7.58 (m, 2H, Ar–H). 13C NMR (100 MHz, DMSO): δ ppm, 172.77 (pyrrolidine 5C=O), 168.42 (pyrrolidine 2C=O), 163.47 (thiazole 2 C), 132.74 (C=CH2) 128.28, 109.09 (aromatic C's), 122.09 (methylidine CH2), 33.93 (pyrrolidine 4 C). TLC (6:4) hexane/EtOAc, Rf = 0.18; GCMS (m/z): 194 (M+). Calcd. for C8H8N2O2S: C, 48.97; H, 4.11; N, 14.28; Found: C, 48.95%; H, 4.13%; N, 14.26%.
2.3.7 1-(1,3-Benzothiazol-2-yl)-3-methylidenepyrrolidine-2,5-dione (4b)
IR (KBr): νmax (cm−1), 3057 (Ar–H), 2968 (aliphatic C–H) 1705 (C=O), 1544 (C=C), 1440 (C=N). 1H NMR (400 MHz, DMSO-d6): δ ppm, 2.89 (s, 2H, CH2), 5.43 (d, 1H, HA, JAB = 2.0 Hz), 6.21 (d, 1H, HB, JAB = 2.0 Hz), 7.17–7.58 (m, 4H, Ar–H). 13C NMR (100 MHz, DMSO): δ ppm, 172.77 (pyrrolidine 5C=O), 168.42 (pyrrolidine 2C=O), 163.47 (thiazole 2 C), 132.74 (C=CH2) 128.28, 127.80, 127.71, 116.34, 116.11, 124.09 (aromatic C's), 122.09 (methylidine CH2), 33.93 (pyrrolidine 4 C). TLC (6:4) hexane/EtOAc, Rf = 0.32; GCMS (m/z): 244 (M+). Calcd. for C12H8N2O2S: C, 59.00; H, 3.30; N, 11.47; Found: C, 58.98%; H, 3.32%; N, 11.45%.
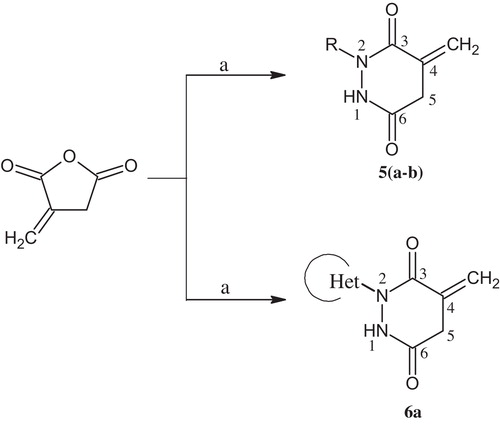
2.4 General procedure for the synthesis of substituted pyridazines 5(a-b), 6a ()
An equimolar mixture of substituted/heterocyclic hydrazide (11.29 mmol) and itaconic anhydride (11.29 mmol) were dissolved in acetic acid (20 mL) and refluxed for 6 h. The completion of the reaction monitored by TLC (Hexane:EtOAc/6:4). After completion of the reaction, the mixture was allowed to cool to room temperature and quenched in ice-cold water under stirring to produce a solid product. The precipitate was filtered and dried, and the crude product was recrystallized using the minimum amount of methanol required to produce a white crystalline powder.
Scheme 2 Synthetic pathways to substituted/heterocyclic pyridazines. (a) Substituted/heterocyclic hydrazide/AcOH, reflux.
2.4.1 5-Methylidene-1-phenyltetrahydropyridazine-3,6-dione (5a)
IR (KBr): νmax (cm−1), 3220 (–NH–), 2997 (Ar–H), 2928 (aliphatic C–H) 1732 (C=O), 1665 (C=C), 1260 (C–N). 1H NMR (400 MHz, DMSO-d6): δ ppm, 3.63 (s, 2H, pyridazine 5CH2), 5.82 (d, 1H, HA, JAB = 1.6 Hz), 6.30 (d, 1H, HB, JAB = 1.6 Hz), 7.16–7.38 (m, 5H, Ar–H), 8.07 (s, 1H, NH). 13C NMR (100 MHz, DMSO): δ ppm, 171.68 (pyridazine 6C=O), 166.04 (pyridazine 3C=O), 139.63 (pyridazine 4 C), 136.88, 130.82, 122.37, 119.14 (aromatic C's), 123.53 (methylidine CH2), 33.28 (pyridazine 5 C). GCMS (m/z): 202 (M+). Calcd. for C11H10N2O3: C, 65.34; H, 4.98; N, 13.85; Found: C, 65.32%; H, 5.00%; N, 13.83%.
2.4.2 1-(2,4-Dinitrophenyl)-5-methylidenetetrahydropyridazine-3,6-dione (5b)
IR (KBr): νmax (cm−1), 3338 (–NH–), 3115 (Ar–H), 2929 (aliphatic C–H) 1735 (C=O), 1620 (C=C), 1498 (N = O). 1H NMR (400 MHz, DMSO-d6): δ ppm, 3.63 (s, 2H, pyridazine 5CH2), 5.85 (d, 1H, HA, JAB = 1.6 Hz), 6.30 (d, 1H, HB, JAB = 1.6 Hz), 7.39–9.32 (m, 3H, Ar–H), 8.27 (s, 1H, NH). 13C NMR (100 MHz, DMSO): δ ppm, 171.69 (pyridazine 6C=O), 167.04 (pyridazine 3C=O), 138.63 (pyridazine 4 C), 146.88, 132.56, 130.82, 127.67, 122.37, 116.14 (aromatic C's), 123.53 (methylidine CH2), 33.28 (pyridazine 5 C). GCMS (m/z): 292 (M+). Calcd. for C11H8N4O6: C, 45.21; H, 2.76; N, 19.17; Found: C, 45.19%; H, 2.78%; N, 19.15%.
2.4.3 1-(1,3-Benzothiazol-2-yl)-5-methylidenetetrahydropyridazine-3,6-dione (6a)
IR (KBr): νmax (cm−1), 3221 (–NH–), 2999 (Ar–H), 2926 (aliphatic C–H) 1734 (C=O), 1668 (C=C), 1261 (C-N). 1H NMR (400 MHz, DMSO-d6): δ ppm, 2.96 (s, 2H, pyridazine 5CH2), 5.43 (d, 1H, HA, JAB = 2.0 Hz), 6.21 (d, HB, JAB = 2.0 Hz), 7.17–7.58 (m, 4H, Ar–H), 7.93 (s, 1H, NH). 13C NMR (100 MHz, DMSO): δ ppm, 173.34 (benzothiozole 2C), 169.02 (pyridazine 6C=O), 165.94 (pyridazine 3C=O), 137.59 (pyridazine 4 C), 133.34, 128.06, 127.47, 126.19, 125.71, 125.50 (aromatic C's), 124.04 (methylidine CH2), 34.01 (pyridazine 5 C). GCMS (m/z): 259 (M+). Calcd. for C12H9N3O2S: C, 55.59; H, 3.50; N, 16.21; Found: C, 55.57%; H, 3.52%; N, 16.19%.
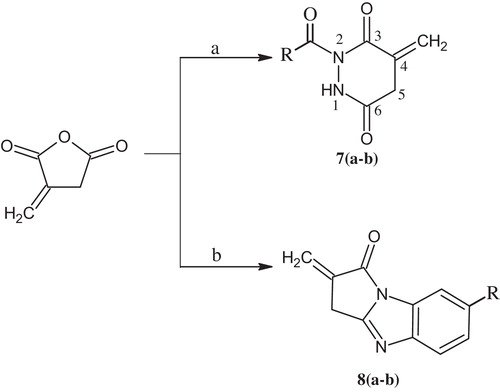
2.5 General procedure for the synthesis of substituted pyridazines 7(a-b) ()
An equimolar mixture of substituted acetohydrazide hydrazide (11.29 mmol) and itaconic anhydride (11.29 mmol) were dissolved in acetic acid (20 mL) and refluxed for 6 h. The completion of the reaction was monitored by TLC (Hexane:EtOAc/6:4). After the completion of the reaction, the mixture was allowed to cool to room temperature and quenched in ice-cold water under stirring to produce a solid product. The precipitate was filtered and dried, and the crude product was recrystallized from the minimum amount of methanol needed to produce a white crystalline powder.
Scheme 3 Synthetic pathways to substituted pyridazines and imidazoles. (a) Substituted acetohydrazide/AcOH, reflux; (b) Substituted o-phenylenediamine, PTSA/toluene, reflux.
2.5.1 1-(Diphenylacetyl)-5-methylidenetetrahydropyridazine-3,6-dione (7a)
IR (KBr): νmax (cm−1), 3246 (–NH–), 3061 (Ar–H), 3026 (aliphatic C–H) 1732 (C=O), 1678 (C=C), 1265 (C–N). 1H NMR (400 MHz, DMSO-d6): δ ppm, 3.28 (s, 2H, pyridazine 5CH2), 5.05 (s, 2H, naphthyl-CH2), 5.60 (d, 1H, HA, JAB = 1.6 Hz), 6.32 (d, 1H, HB, JAB = 1.6 Hz), 7.20–7.57 (m, 10H, Ar–H), 7.77 (s, 1H, NH). 13C NMR (100 MHz, DMSO): δ ppm, 170.74 (pyridazine 6C=O), 167.70 (CH2–C=O), 162.79 (pyridazine 3C=O), 136.89 (pyridazine 4 C), 136.00, 130.61, 129.07, 127.15, (aromatic C's), 123.85 (methylidine CH2), 51.63 (naphthyl-CH2), 32.59 (pyridazine 5C). GCMS (m/z): 320 (M+). Calcd. for C19H16N2O3: C, 71.24; H, 5.03; N, 8.74; Found: C, 71.22%; H, 5.05%; N, 8.72%.
2.5.2 5-Methylidene-1-(naphthalen-2-ylacetyl)tetrahydropyridazine-3,6-dione (7b)
IR (KBr): νmax (cm−1), 3221 (–NH–), 2999 (Ar–H), 2926 (aliphatic C–H) 1734 (C=O), 1668 (C=C) 1261 (C-N). 1H NMR (400 MHz, DMSO-d6): δ ppm, 3.29 (s, 2H, pyridazine 5CH2), 4.15 (s, 2H, naphthyl-CH2), 5.59 (d, 1H, HA, JAB = 1.6 Hz), 6.31 (d, 1H, HB, JAB = 1.6 Hz), 7.19–7.58 (m, 7H, Ar–H), 7.77 (s, 1H, NH). 13C NMR (100 MHz, DMSO): δ ppm, 169.74 (pyridazine 6C=O), 166.70 (CH2–C=O), 161.79 (pyridazine 3C=O), 134.00 (pyridazine 4C), 131.89, 130.61, 129.07, 128.99, 128.81, 128.70, 127.15, 126.52, 125.65, 122.98 (aromatic C's), 123.85 (methylidine CH2), 39.63 (naphthyl-CH2), 32.59 (pyridazine 5C). GCMS (m/z): 294 (M+). Calcd. for C17H14N2O3: C, 69.38; H, 4.79; N, 9.52; Found: C, 69.36%; H, 4.81%; N, 9.50%.
2.6 General procedure for the synthesis of substituted imidazole 8(a-b) ()
One-hundred millilitres of toluene and 100 mg of p-toluene sulfonic acid (PTSA) were added to an equimolar mixture of itaconic anhydride (6.0 mmol) and o-phenylenediamine (6.0 mmol) in a reflux apparatus equipped with a Dean-Stark trap. The reaction was heated to 110 °C for 3–4 h, and the completion of reaction was monitored by TLC (Hexane:EtOAc/6:4). After the completion of the reaction, toluene was removed under vacuum conditions and substituted imidazole was isolated by column chromatography over silica gel using DCM as an eluent.
2.6.1 2-Methylidene-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-1-one (8a)
IR (KBr): νmax (cm−1), 3057 (Ar–H), 2964 (aliphatic C–H) 1699 (C=O), 1544 (C=C), 1444 (C=N). 1H NMR (400 MHz, DMSO-d6): δ ppm, 3.04 (s, 2H, CH2), 5.75 (d, 1H, HA, JAB = 1.2 Hz), 6.15 (d, 1H, HB, JAB = 1.2 Hz), 7.30–7.52 (m, 4H, Ar–H). 13C NMR (100 MHz, DMSO): δ ppm, 189.15 (N–C=O), 149.98 (C=CH2) 139.17, 138.47, 137.34, 124.26, 120.82 (aromatic C's), 130.15 (methylidine CH2), 24.16 (CH2). TLC (6:4) hexane/EtOAc, Rf = 0.26; LCMS (m/z): 185 (M++1). Calcd. for C11H8N2O: C, 71.73; H, 4.38; N, 15.21; Found: C, 71.71%; H, 4.40%; N, 15.19%.
2.6.2 2-Methylidene-7-(phenylcarbonyl)-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-1-one (8b)
IR (KBr): νmax (cm−1), 3057 (Ar–H), 2970 (aliphatic C–H) 1697 (C=O), 1544 (C=C), 1444 (C=N). 1H NMR (400 MHz, DMSO-d6): δ ppm, 2.14 (s, 2H, CH2), 5.77 (d, 1H, HA, JAB = 1.2 Hz), 6.18 (d, 1H, HB, JAB = 1.2 Hz), 7.39–8.01 (m, 8H, Ar–H). 13C NMR (100 MHz, DMSO): δ ppm, 195.95 (C=O), 189.15 (N–C=O), 150.98 (C=CH2) 139.15, 138.47, 138.34, 132.70, 132.62, 132.30, 125.22, 124.26, 120.82, 116.14, 115.62 (aromatic C's), 129.95 (methylidine CH2), 23.16 (CH2). TLC (6:4) hexane/EtOAc, Rf = 0.41; LCMS (m/z): 289 (M++1). Calcd. for C18H12N2O2: C, 74.99; H, 4.20; N, 9.72; Found: C, 74.97%; H, 4.22%; N, 9.70%.
2.7 Pharmacology
2.7.1 1,1-Diphenyl-2-picrylhydrazyl (DPPH) radical scavenging assay
The free radical scavenging activity of the compounds was measured using a previously described method with DPPH radicals [Citation10]. The ability to scavenge DPPH radicals was calculated from the following equation:where A0 is the absorbance of the control reaction and A1 is the absorbance of the samples or standards. Each sample was assayed at 1 mg/mL, and all experiments were carried out in triplicate.
2.7.2 Ferric reducing power assay (FRAP)
The reducing power of the synthesized compounds was determined according to a previously described method [Citation11]. All experiments were carried out in triplicate, and the reducing power assay was used an effective concentration (mg/mL) with a glutathione absorbance of 0.5.
2.7.3 Nitric oxide scavenging activity
Nitric oxide radical scavenging was estimated based on the Griess Illosvoy reaction [Citation12].
2.7.4 Antimicrobial activity (disc diffusion method)
Microbial isolates: the microorganisms used for the microbial sensitivity assay were Gram-positive Staphylococcus aureus (NCIM 2079) and Bacillus subtilis (ATCC 6633) and Gram-negative Pseudomonas aeruginosa (NCIM 2200) and Klebsiella pneumoniae (NCIM 2957). All of the bacterial strains were procured from the National Chemical Laboratory of Pune, India. The fungal strain viz. Candida albicans MTCC No. 227 was obtained from IMTECH, (Chandigarh, India). The bacterial strains were maintained on nutrient agar slants, and the fungal strains were maintained on potato dextrose agar slants.
Antibacterial and antifungal assays were carried out by the disc diffusion method [Citation13]. Streptomycin and Nystatin (HIMEDIA) were used as positive standards against the bacterial and fungal strains, respectively.
2.7.5 Minimum inhibitory concentration (MIC)
The MIC of the DMSO extracts was determined by using different concentrations of extracts in Mueller-Hinton Broth for bacteria and Potato Dextrose Broth for fungi by the macro dilution method [Citation14,Citation15]. The lowest concentration of DMSO extract that was needed to inhibit the visible growth of microorganisms was considered to be the MIC.
2.7.6 Molecular docking studies
The docking analysis to assess the antioxidant activity of screened compounds 5a, 5b, 6a, 8a, and 8b was carried out with tyrosinase using the HEX 6.3 docking software. Rigid body docking was performed with HEX 6.3 [Citation16] using an SP Fourier Transform, FFT steric scan, FFT final search and MM refinement.
The tyrosinase enzyme structure (PDB ID: 3NM8) of Bacillus megaterium was downloaded from the PDB [www.pdb.org/pdb]. The enzyme structure containing hetero molecules was removed before the docking was performed. The corresponding CIF files of the derivatives were converted into PDB files using ArgusLab. These PDB structures were used for the docking studies with HEX software [Citation16]. The ligand proves to be the best inhibitor because the ligand and receptor dock together with a higher energy of binding (E-total value).
3 Results and discussions
3.1 Chemistry
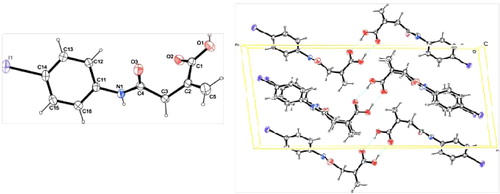
Substituted/heterocyclic imides 3(a-e) and 4(a-b) were synthesized through the cyclization of intermediate substituted/heterocyclic oxobutanoic acid 1(a-e) and 2(a-b), as shown in . In first step, the nucleophilic reagents attack the γ-carbonyl group with the formation of γ-oxobutanoic acids. Similar derivatives to intermediate oxobutanoic acids were synthesized and characterized by our group. Their biological activities were also evaluated by our research group [Citation17–Citation19]. The intermediates reported here are new except for 4-[(4-iodophenyl)amino]-2-methylidene-4-oxobutanoic acid (1d), the crystal X-ray data of which () are shown [Citation20]. Based on the crystal study of compound 1d, we found that there is a twist between the mean planes of the methylidene group and the amide group to an extent of approximately 100°. The presence of a weak interaction between the methylidene and amide groups supports this conformation. This arrangement could be favoured when the carboxylic acid group prefers to stay away from the mean plane (the trans form). If the carboxylic acid group stays in the cis position, it leads to a hindered conformation. Hence, it can be concluded that the Z (cis) form of the molecules 1(a-e) and 2(a-b) becomes unstable, thus favouring the E (trans) form.
Fig. 1 The molecular structure and molecular packing of intermediate compound 1d with displacement ellipsoids drawn at the 50% probability level.
The intermediate compounds 1(a-e) and 2(a-b) undergo cyclization in the presence of NaOAc to yield substituted/heterocyclic imides 3(a-e), 4(a-b). The imide structures were established based on analytical and spectral (IR, 1H & 13C NMR, GCMS/LCMS) data. In the IR spectra of substituted/heterocyclic oxobutanoic acid, 1(a-e) and 2(a-b) exhibited stretching bands in the region of 3111–3285 cm−1. These were attributed to the amide NH, which was missing in the IR spectra of the substituted/heterocyclic imides. This suggested the formation of imides from intermediate oxobutanoic acids. The 1H NMR spectrum of 3a showed a singlet signal at δ 3.54 ppm due to an imide proton at C-4. The two doublets that appeared at δ 5.78 ppm and δ 6.31 ppm were due to HA and HB of the exocyclic methylidine protons. The coupling constant (J) of HA and HB was 2.0 Hz, which was characteristic of germinal coupling in the methylidene group. The signals for aromatic protons appeared as multiplets in the range of δ 7.17–7.38 ppm. The 13C NMR showed peaks at δ 172.77 ppm and δ 169.42 ppm due to the carbonyl groups at the second and fifth position of the pyrrolidine ring, respectively. The signal at δ 132.74 ppm was due to the C=CH2 group of the pyrrolidine ring. All other signals appearing in the 13C NMR spectrum accounted for the carbon atoms in the molecule. Further mass spectra showed (M+), (M++1) and (M++2) peaks, confirming the presence of halogens in compounds 3(a-e) and 4(a-b).
The reaction of itaconic anhydride with different substituted aryl/heterocyclic hydrazides and acid hydrazides yielded the 2-substituted aryl/heterocyclic pyridazine derivatives 5(a-b), 6a, and 7(a-b), as shown in Schemes and . The pyridazine structures were established based on analytical and spectral (IR, 1H & 13C NMR, GCMS/LCMS) data.
The IR spectra of the 2-substituted aryl/heterocyclic pyridazine derivatives 5(a-b), 6a, 7(a-b) showed stretching bands of approximately 3220–3338 cm−1 and 1732–1735 cm−1 due to the NH and carbonyl groups, respectively. The 1H NMR spectrum of 5a showed a singlet at δ 3.63 ppm, corresponding to pyridazine protons at C-5, and another singlet signal at δ 8.07 ppm, corresponding to the NH proton. Together, these signals confirmed the formation of pyridazine. The two doublets appeared at δ 5.82 ppm and δ 6.30 ppm due to the HA and HB of the exocyclic methylidine protons. The coupling constant (J) for HA and HB was 1.6 Hz, which is characteristic of the germinal coupling of the methylidene group. In 13C NMR, peaks observed at δ 171.68 ppm and δ 166.04 ppm were attributed to the pyridazine carbonyl groups C-6 and C-3, respectively. In contrast, C-4 and C-5 were responsible for the remaining pyridazine carbon peaks at δ 139.63 ppm and δ 33.28 ppm, respectively. The number of signals that appeared in the 13C NMR spectrum accounted for the number of carbon atoms present in the molecule. Mass spectra exhibited molecular ion peaks m/z at (M+) for compounds 5(a-b), 6a, and 7(a-b). Similarly, as shown in , the synthesis of substituted imidazole derivatives 8(a-b) was carried out in the presence of the PTSA catalyst. Based on the analytical and spectral (IR, 1H & 13C NMR, GCMS/LCMS) data, the structure of the imidazoles was established. The characterization data of all of the synthesized products (1(a-e), 2(a-b), 3(a-e), 4(a-b), 5(a-b), 6a, 7(a-b), and 8(a-b)) are summarized in .
Table 1 Characterization data of compounds 3(a-e), 4(a-b), 5(a-b), 6a, 7(a-b), and 8(a-b).
3.2 Pharmacology
3.2.1 DPPH radical scavenging assay
The DPPH assay used here was based on a previously reported method [Citation13]. The radical scavenging mechanism is based on the transfer of the acidic H-atom from the compound to the DPPH radical to form DPPH-H. Each sample was assayed at 1 mg/mL, and the % RSC data are presented in .
Fig. 2 Comparison of the DPPH scavenging activity of newly synthesized ITA derivatives 3(a-e), 4(a-b), 5(a-b), 6a, 7(a-b), and 8(a-b).
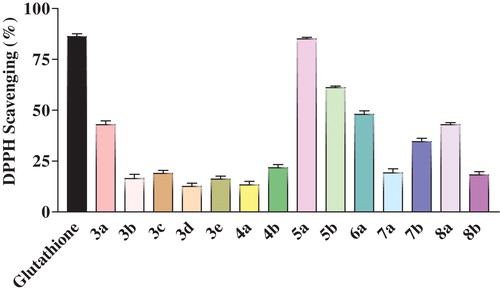
Of the tested compounds, 1 mg/mL 5-methylidene-1-phenyltetrahydropyridazine-3,6-dione (5a) showed a better radical scavenging capacity than the standard glutathione. This enhanced activity could be due to the presence of acidic protons in its pyridazine moiety [Citation21]. The compounds 3a, 6a, and 8a also showed moderate activity due to the presence of imide, pyridazine and imidazole functionalities in these molecules.
3.2.2 Ferric reducing antioxidant power (FRAP) assay
The reducing capacity of a compound may serve as a significant indicator of its potential antioxidant activity [Citation22]. Compounds with reducing power give up electrons and can reduce the oxidized intermediates of lipid peroxidation processes so that they can act as primary and secondary antioxidants [Citation23].
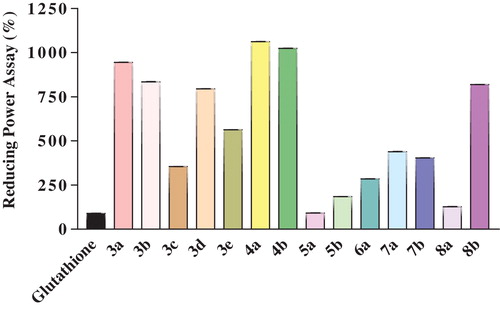
The reducing power assay is expressed in an effective concentration (mg/mL) equivalent of 0.5 glutathione absorbance. Many of the tested compounds showed significant reducing power capacities. Compared to glutathione, 5-methylidene-1-phenyltetrahydropyridazine-3,6-dione (5a) and 2-methylidene-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-1-one (8a) showed good reducing capacities, while compounds 3c and 6a exhibited more moderate reducing capacities. Although the reducing power of these compounds might be due to the presence of imide, pyridazine and imidazole functionalities, their net activity could depend on the different substituents present in the molecule ().
Fig. 3 Comparison of the reducing power capacity of newly synthesized ITA derivatives 3(a-e), 4(a-b), 5(a-b), 6a, 7(a-b), and 8(a-b).
3.2.3 Nitric oxide scavenging assay
Active oxygen species and free radicals are involved in a variety of pathological events. In addition to ROSs, inflammation, cancer and other pathological conditions are also related to nitric oxide. In the present study, nitric oxide (NO•) was generated from sodium nitroprusside in an aqueous solution at physiological pH, which interacts with oxygen to produce nitrate ions. This process can be estimated using the Greiss reagent [Citation24]. The NO scavenging effect of the synthesized compounds is represented in .
Fig. 4 Comparison of the nitric oxide scavenging activity of newly synthesized ITA derivatives 3(a-e), 4(a-b), 5(a-b), 6a, 7(a-b), and 8(a-b).
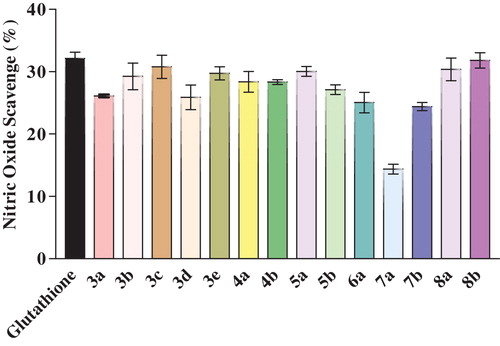
Of the tested compounds, 5-methylidene-1-phenyltetrahydropyridazine-3,6-dione (5a), 2-methylidene-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-1-one (8a) and 2-methylidene-7-(phenylcarbonyl)-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-1-one (8b) showed good NO scavenging ability at a concentration of 1 mg/mL in comparison with the standard glutathione. Such enhanced activity could be due to the presence of a pyridazine and imidazole moiety [Citation21,Citation25]. Compounds 3b, 3c, 3e, 4a, and 4b also showed moderate activity due to the presence of imide functionality.
3.2.4 Correlation of antioxidant activities
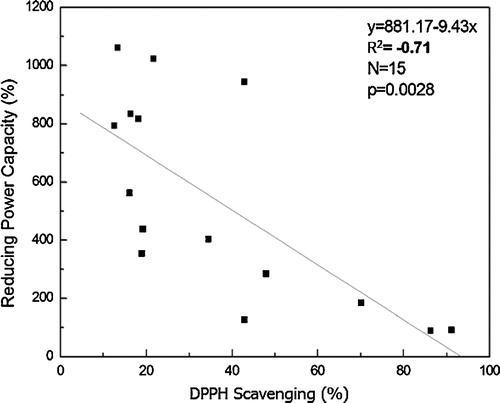
The correlation between the antioxidant activities of the DPPH radical scavenging assay, reducing power assay (FRAP) and nitric oxide scavenging assay was tested. Linear regression analyses between DPPH radical scavenging and the reducing power assay showed a significant negative correlation value, with a R2 value 0.71 (), which indicated that the higher the DPPH activity of the compound, the lower the value of FRAP and the higher the antioxidant activity. The correlation between the DPPH radical scavenging assay and the nitric oxide scavenging assay and between the reducing power assay and the nitric oxide scavenging assay were poor, with R2 < 0.4.
Fig. 5 Linear correlation between the DPPH and the reducing power assay.
3.2.5 Antimicrobial activity
The newly synthesized compounds were screened for their antibacterial and antifungal activities against four bacterial strains and one fungal strain. The values of the resulting zones of inhibition are summarized in .
Table 2 Antibacterial and antifungal activities of the synthesized compounds 3(a-e), 4(a-b), 5(a-b), 6a, 7(a-b), and 8(a-b).
The compound 1-(4-iodophenyl)-3-methylidenepyrrolidine-2,5-dione (3d) was found to be most active against S. aureus, B. subtilis and Klebsiella pneumonia, with an inhibition zones of 21.33 mm, 24.66 mm and 21.67 mm, respectively, at an MIC of 50 μg/L. The compounds 3a, 3b, 3e, and 7b were found to be active against some of the bacterial strains. The high activity of 3d could be attributed to the presence of the imide moiety, containing the 4-iodo phenyl substituent [Citation26]. In addition, the substituted imide moiety (3a, 3b, 3e) and pyridazine group (7b) may have helped to inhibit microorganism growth. shows the minimum inhibitory concentrations (MIC) values of the active antibacterial compounds.
Table 3 MIC data for the active antimicrobial compounds.
Compounds 1-(4-chlorophenyl)-3-methylidenepyrrolidine-2,5-dione (3a) and 3-methylidene-1-(1,3-thiazol-2-yl)pyrrolidine-2,5-dione (4a) were found to be most active against C. albicans, with an inhibition zone of 21.33 mm at an MIC of 50 μg/L and 100 μg/L, respectively (). Although compounds 3b and 3d showed moderate activity against the selected fungal strain, they exhibited lower potencies than the standard drug. This finding suggests that different functionalities present in the molecules contribute to antifungal activities in different ways.
3.2.6 Molecular docking studies
Molecular docking studies allow for the virtual screening of a database of compounds to predict the strongest binders based on various scoring functions. This method explores the ways in which two molecules, such as a drug and an enzyme tyrosinase receptor, fit together like pieces of a three-dimensional jigsaw puzzle. The molecules bind to the receptor to inhibit its function, thus acting as drugs. Docking results between the screened heterocyclic derivatives and selected receptor, i.e., the tyrosinase (PDB ID: 3NM8) enzyme from B. megaterium, are tabulated in . The structures of the receptor, ligand, and ligand–receptor complex are shown in –. As shown in , the ligand–receptor fits has the best fit (the highest binding energy) for compounds 2-methylidene-7-(phenylcarbonyl)-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-1-one (8b) (energy value −221.41), followed by 2-methylidene-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-1-one (8a) (energy value −213.96) and 1-(1,3-benzothiazol-2-yl)-5-methylidenetetrahydropyridazine-3,6-dione (6a) (energy value -203.69). 5-methylidene-1-phenyltetrahydro pyridazine-3,6-dione (5a) showed the lowest binding energy, with the highest total energy value of −153.70. Docking studies showed that Tyr267A, Pro268A, Ala44A, Tyr267B, and Pro268B in the tyrosinase enzyme (PDB ID: 3NM8) likely play a major role in substrate binding or catalysis. These in silico study results are in line with the in vitro enzyme assay results and support the overall outcome of this study.
Fig. 6 Structure of the tyrosinase enzyme (PDB ID: 3NM8) from Bacillus megaterium.
Fig. 7 The docking result showing the position of compound 5a inside Bacillus megaterium tyrosinase.
Fig. 8 The docking result showing the position of compound 5b with the nearby amino acid groups of Bacillus megaterium tyrosinase.
Fig. 9 The position of compound 6a on Bacillus megaterium tyrosinase after docking with the Hex docking software.
Fig. 10 The docking result showing the position of compound 8a with a nearby amino acid group of Bacillus megaterium tyrosinase.
Fig. 11 The position of compound 8b on Bacillus megaterium tyrosinase after docking with the HEX software.
Table 4 Docking study results of the screened antioxidant compounds.
3.2.7 Structure activity relationship (SAR)
Of the compounds tested with the DPPH radical scavenging assay, the reducing power assay (FRAP) and the nitric oxide scavenging assay, the imidazole- and pyridazine-derived compounds were found to be the most active, with a competitive edge over the imide-derived compounds. Of the benzimidazole derivatives, compound 8a (R=H) showed more activity than compound 8b (R=C6H5–CO). Likewise, the phenyl-substituted pyridazine derivative 5a exhibited good activity compared to that of 5b, containing an electron-withdrawing 2,4-dinitro phenyl substituent on the pyridazine moiety. The results of the molecular docking study supported the results of the in vitro antioxidant assays. Of the compounds that docked with tyrosinase (PDB ID: 3NM8), compounds 8a and 8b were found to be the most potent molecules, with binding energies with minimum total energy values of −213.96 and −221.41, respectively. The benzimidazol-1-ones 8(a-b) showed better activity than the pyrrolidine-2,5-diones 3(a-e) and 4(a-b) and the pyridazine-3,6-diones 5(a-b) and 6a.
4 Conclusion
New heterocyclic derivatives were prepared by a simple, high-yield, one-/two-step process from an ITA precursor. All of the compounds were well-characterized by spectral and analytical data. Compounds 5-methylidene-1-phenyltetrahydro pyridazine-3,6-dione (5a) and 2-methylidene-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-1-one (8a) exhibited good DPPH radical scavenging activity, reducing the power capacity and nitric oxide scavenging activity in comparison with the standard glutathione. Linear regression analyses between the DPPH radical scavenging and reducing power assay showed a significant negative correlation, with R2 equal to 0.71. 1-(4-iodophenyl)-3-methylidenepyrrolidine-2,5-dione (3d) showed good antibacterial activity, whereas 1-(4-chlorophenyl)-3-methylidenepyrrolidine-2,5-dione (3a) and 3-methylidene-1-(1,3-thiazol-2-yl)pyrrolidine-2,5-dione (4a) exhibited good antifungal properties. Among the compounds that docked with tyrosinase (PDB ID: 3NM8), 2-methylidene-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-1-one (8a) and 2-methylidene-7-(phenylcarbonyl)-2,3-dihydro-1H-pyrrolo[1,2-a]benzimidazol-1-one (8b) exhibited higher binding compared to other molecules. In conclusion, all of the newly synthesized compounds exhibited moderate to good activities.
Acknowledgements
B.N. thanks the UGC grant no. (SR/S9/Z-23/2010/32 dated 15/03/2011) for financial assistance through a one-time BSR grant (F.4-10/2010(BSR) dated July 2011) for the purchase of chemicals. P.S.N. thanks DST-PURSE grant no. (F.4-10/2010 (BSR) dated July 2011) for financial assistance.
Notes
Peer review under responsibility of Taibah University.
References
- B.N.AmesM.K.ShigenagaT.M.HagenOxidants, antioxidants and the degenerative diseases of agingProc. Natl. Acad. Sci. U. S. A.90199379157922
- D.CostaA.GomesJ.L.F.C.LimaE.FernandesSinglet oxygen scavenging activity of non-steroidal anti-inflammatory drugsRedox Rep.132008153
- D.CostaA.VieiraE.FernandesDipyrone and aminopyrine are effective scavengers of reactive nitrogen speciesRedox Rep.112006136142
- A.PekselC.CelikN.OcalR.YanardagAntioxidant and radical scavenging activities of some norcantharidin and bridged perhydroisoindole derivativesJ. Serb. Chem. Soc.7820131525
- E.B.CaliskanM.SukurogluT.CobanE.BanogluS.SuzenScreening and evaluation of antioxidant activity of some pyridazine derivativesJ. Enzym. Inhib. Med. Chem.232008225229
- S.De StrayoJ.AdhikariV.P.Tilak-JainT.P.A.MenonDevasagayamAntioxidant activity of an aminothiazole compound: possible mechanismsChem. Biol. Interact.1732008215223
- C.KusG.Ayhan-KılcıgilM.TunçbilekN.AltanlarT.ÇobanB.Can-EkeM.IscanAntimicrobial and antioxidant activities of some benzimidazole derivativesLett. Drug Des. Discov.62009374379
- S.J.S.FloraStructural, chemical and biological aspects of antioxidants for strategies against metal and metalloid exposureOxid. Med. Cell. Longev.22009191206
- P.S.NayakB.NarayanaB.K.SarojiniK.HegdeK.S.ShashidharaDesign and synthesis of novel heterocyclic acetamide derivatives for potential analgesic, anti-inflammatory and antimicrobial activitiesMed. Chem. Res.201410.1007/s00044-014-1003-3
- M.S.BloisAntioxidant determinations by the use of a stable free radicalNature181195811991200
- M.OyaizuStudies on products of the browning reaction. Antioxidative activities of browning reaction products prepared from glucosamineJpn. J. Nutr.441986307315
- D.C.GarrattThe Quantitative Analysis of Drugsvol. 31964Chapman and Hall Ltd.Japan456458
- G.Vardar-UnluF.CandanSokemenD.DaferraM.PollissiouM.SokemenAntimicrobial and antioxidant activity of the essential oil and methanol extracts of Thymus pectinatus Fisch. et Mey. Var. pectinatusJ. Agric. Food Chem.5120036367
- National Committee for Clinical Laboratory Standards (NCCLS)Performance Standard for Antimicrobial Disc Susceptibility TestApproved standard 7th ed.2000M2-A7 NCCLS documentWayne Pa
- K.O.AkinyemiO.OladapoC.E.OkwaraC.C.IbeK.A.FasureScreening of crude extracts of six medicinal plants used in south-west Nigerian unorthodox medicine for anti-methicillin resistant Staphylococus aureus activityBMC Complement. Alter. Med.520056
- D.W.RitchieEvaluation of protein docking predictions using Hex 3.1 in CAPRI rounds 1 and 2Proteins200398106
- P.S.NayakB.NarayanaS.AnthalV.K.GuptaR.KantH.S.YathirajanSynthesis, characterization, crystal structure and thermal analysis of 4-[(3-acetylphenyl)amino]-2-methylidene-4-oxobutanoic acidMol. Cryst. Liq. Cryst.5922014249258
- P.S.NayakB.NarayanaB.K.SarojiniSynthesis, characterization and in vitro biological evaluation of 4-[(1,5-dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)amino]-2-methylidene-4-oxobutanoic acidIndones. Am. J. Pham. Res.4201418811888
- P.S.NayakB.NarayanaJ.P.JasinskiH.S.YathirajanM.Kaur4-(3-Fluoro-4-methyl anilino)-2-methylidene-4-oxobutanoic acidActa Cryst.E692013o1752
- P.S.NayakB.NarayanaH.S.YathirajanT.GerberB.Van BrechtR.Betz4-(4-Iodo anilino)-2-methylene-4-oxobutanoic acidActa Cryst.E692013o83
- B.Çalişkan-ErgünM.şüküroğluT.CobanE.BanoğluS.SuzenScreening and evaluation of antioxidant activity of some pyridazine derivativesJ. Enzym. Inhib. Med. Chem.232008225229
- M.OktayI.GulcinO.I.KufreviogluDetermination of in vitro antioxidant activity of fennel (Foeniculum vulgare) seed extractsLWT Food Sci. Technol.362003263271
- S.ChandaR.DaveIn vitro models for antioxidant activity evaluation and some medicinal plants possessing antioxidant properties: an overviewAfr. J. Microbiol. Res.32009981996
- J.A.WallachS.J.HuangCopolymers of itaconic anhydride and methacrylate-terminated poly(lactic acid) macromonomersBiomacromolecules12000174179
- M.SureshP.LavanyaC.V.RaoSynthesis, antibacterial, antifungal and antioxidant activity studies on 2-benzylthio- and 2-benzylsulfonyl-1H-imidazolesDer Pharmacia Lettre22010393402
- X.Jiang 1W.LiuW.ZhangF.JiangZ.GaoH.ZhuangL.FuSynthesis and antimicrobial evaluation of new benzofuran derivativesEur. J. Med. Chem.46201135263530
Appendix A
Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.jtusci.2014.09.005.