
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
As a continuation of our research on the development and optimization of the biological activities/proprieties of acridine derivatives, a series of 31 molecules based on 9-aniliioacridines (25 training set and 6 test set) were subjected to 3D quantitative structure propriety relationship QSPR analyses for their drug-DNA binding proprieties using multiple linear regression (MLR) and multiple non-linear regression (MNLR). Quantum chemical calculations using density functional theory (B3LYP/6-31G (d) DFT) methods was performed on the studied compounds and used to calculate the electronic and quantum chemical parameters.
The models were used to predict the association constant of the DNA drug binding of the test set compounds, and the agreement between the experimental and predicted values was verified. The descriptors determined by QSPR studies were used for the study and design of new compounds. The statistical results indicate that the predicted values were in good agreement with the experimental results (r = 0.935 and r = 0.936 for MLR and MNLR, respectively). To validate the predictive power of the resulting models, the external validation multiple correlation coefficients were 0.932 and 0.939 for the MLR and the MNLR, respectively. These results show that both models possess a favourable estimation stability and good prediction power.
1 Introduction
The ease of synthesis, attractive colouration and crystallinity of acridine derivatives has long attracted the attention of medicinal chemists. The acridine family includes a wide range of planar tri-cyclic aromatic molecules with various biological properties and consists of a nitrogen atom (N-atom) in its heterocyclic nucleus. The natural and synthetic compounds of the acridine family are well known therapeutic agents due to their wide range of pharmacological and biological activities, including anti-leishmanial [Citation1,Citation2], anti-microbial [Citation3], anti-oxidant [Citation4], anti-malarial [Citation5], anti-inflammatory [Citation6], analgesic [Citation7], anti-parasitic [Citation8], anti-tumoural [Citation9], anti-bacterial or anti-cancer chemotherapy [Citation10–Citation13] activities, among others.
The diversity of the biological and pharmacological activities has given acridines a respectable reputation in chemotherapy in the 20th century [Citation14].
One of the important phenomena of DNA is their ability to reversibly bind planar molecules that can be inserted between the base pairs of the double helix [Citation15]. Acridines are fused linear tri-cyclic aromatic molecules with planar geometry that bind tightly, but reversibly, to DNA by intercalating between adjacent base pairs [Citation16,Citation17]. The driving force for this binding comes primarily from stacking interactions between the acridine nucleus and the DNA bases and is sufficiently large to physically unwind the DNA double helix to accommodate the inserted ligand. The majority of the biological effects of acridine derivatives are considered to result from this mode of non-covalent interactions with DNA [Citation18].
Generally, hetero-aromatic molecules bind to DNA by intercalating (i.e., a non-covalent interaction in which the drug is held rigidly and perpendicular to the helix axis) and stacking between the base pairs of the double helix. The principal driving forces for the intercalation are stacking and charge-transfer interactions, but hydrogen bonding and electrostatic forces also play a role in stabilization [Citation19].
9-Anilinoacridines, as intercalators of double-stranded (duplex) DNA, have been explored extensively as antitumour agents. In particular, m-AMSA and CI-921 are, in fact, used clinically for the treatment of leukaemia [Citation20–Citation23].
To discover new active antitumour compounds, we examined the DNA-ethidium fluorescence quenching effect of these compounds and found a substance that exhibits a stronger fluorescence quenching effect than m-ASMA. Moreover, it has been established that the fluorescence quenching demonstrates a very good correlation with antitumour activity [Citation24–Citation26].
In an investigation of the structure propriety relationships in the (AMSA) tumour inhibitory analogues, the DNA binding properties of a series of 9-aniliioacridines was determined by drug competition with the fluorochrome ethidium for available sites. The decrease in fluorescence of a DNA-ethidium complex by the addition of a drug is due to both the drug displacement of the bound ethidium and the quenching of the fluorescence of the bound ethidium by the bound drug. The measurement of both factors allows the drug-DNA association constants (K) to be determined [Citation27,Citation28].
The experiment is a direct method of obtaining the activity/propriety data of organic compounds. However, this approach suffers from many deficiencies, including the requirement of myriads of trial organisms, high cost, long period of time, significant variations in the measured values between laboratories, and so on. Consequently, it would be impossible to determine the drug-DNA association constants of all of the organic compounds by experimentation. As new compounds are emerging, other difficulties will follow. Therefore, it is necessary to use theoretical research to compensate for the disadvantages of experimentation and to predict the data for compounds quickly and precisely. In this research paper, we focus on the drug-DNA binding constants of some acridine derivatives.
With the rapid development of computer science and theoretical quantum chemical studies, the quantum chemical parameters of compounds can be obtained quickly and precisely by computation. These structural parameters, along with the introduction of quantitative structure activity/propriety relationship (QAPR/QSPR) models, can increase the interpretability and predictability of the activities/proprieties of new organic compounds.
In this work, we attempt to establish a quantitative structure propriety relationship for the association constant (K) of drug binding to DNA by studying a series of 31 substituted 9-aniliioacridine derivatives. We accordingly propose quantitative models and try to interpret the propriety of the compounds relying on multivariate statistical analyses. Thus, we can predict the association constant (K) for drug binding to DNA.
2 Material and methods
2.1 Experimental data
To determine a quantitative structure function relationship, we studied a series of 31 selected 9-aniliioacridine derivatives that were synthesized and that had their antitumour activity evaluated by Bruce et al. [Citation27]. Twenty-five molecules were selected to propose the quantitative model (training set) as well as 6 compounds that were not used in the training set were selected randomly served to test the performance of the proposed model (test set). In reality, Bruce et al. proposed 65 compounds; the remaining compounds had structures that differed from the structures required for this study.
shows the chemical structures of the studied compounds, and the experimental association constants for the drug binding to DNA (the concentration of drug needed to displace 50% of the ethidium) of the studied compounds were taken from Ref. [Citation27] ().
Fig. 1 Chemical structure of the studied compounds.
Table 1 Observed Log K for the 9-aniliioacridine derivatives.
2.2 Computational methods
An attempt has been made to correlate the propriety of these compounds with various physicochemical parameters. DFT (density functional theory) and TD-DFT methods were used in this study. 3D structures of the molecules were generated using the Gauss View 3.0, and then, all of the calculations were performed using the Gaussian 03 W program series. Geometry optimization of the 31 compounds was carried out by a B3LYP function employing a 6–31G (d) basis set [Citation29,Citation30]. The geometry of all of the species under investigation was determined by optimizing all of the geometrical variables without any symmetry constraints [Citation31].
2.3 Calculation of the molecular descriptors
From the results of the DFT calculations, the quantum chemistry descriptors were obtained for the model building as follows: the total energy, ET (eV); highest occupied molecular orbital energy, EHOMO (eV); lowest unoccupied molecular orbital energy, ELUMO (eV); difference in absolute value Gap (eV); dipole moment, μ (Debye); absolute hardness, η (eV); absolute electronegativity, χ (eV); electrophilicity index, ω (eV); and sum of the negative charges on the molecule (TNC) were deduced from the stable structure of the neutral form. η, χ and ω were determined from [Citation32]:
The transition energies were calculated in the ground-state with excited-state geometries using TD-DFT calculations on the fully optimized geometries. The results obtained gave us the absorption maximum, λmax (nm), their corresponding activation energy, Ea (eV), and the factor oscillation strengths, S.O.
2.4 Statistical analysis
To explain the structure-activity relationship, these 12 descriptors were calculated for the 31 molecules using the Gaussian 03W and Gauss View software. The study that we conducted consists of multiple linear regression (MLR) and non-linear regression (MNLR), which are available in the XLSTAT software [Citation33].
The multiple linear regression statistical technique is used to study the relationship between one dependent variable and several independent variables. It is a mathematical technique that minimizes the differences between actual and predicted values. It has also served to select descriptors that are used as input parameters in multiple non-linear regression (MNLR).
The MLR and MNLR techniques were used to predict the association constant for drug binding to DNA values, Log(K). The equations were justified by the correlation coefficient (r), the Mean Squared Error (MSE), the Fishers F-statistic (F), and the significance level (F-value) [Citation34].
3 Results and discussion
3.1 Data set for analysis
The QSPR analysis was performed using the experimental association constant for drug binding to DNA values for the 31 selected molecules as reported by Bruce et al. [Citation27]; the values of the 12 chemical descriptors are shown in .
Table 2 Values of the parameters obtained by DFT/TD-DFT calculation for the training set.
3.2 Multiple linear regression (MLR)
To propose a mathematical model and to quantitatively evaluate the substituent's physicochemical effects on log K for the entire set consisting of 31 molecules, we submitted the data matrix that was composed of the 12 variables that corresponded to the 25 molecules (training set) to a descendent multiple regression analysis.
The decreasing study of MLR based on the elimination of descriptors aberrant until a valid model (including the critical probability: p-value < 0.05 for all descriptors and the model complete). This method used the coefficients r, r2, MSE and F-values to select the best regression performance, where r is the correlation coefficient; r2 is the coefficient of determination; MSE is the mean squared error; and F is the Fisher F-statistic.
Treatment with multiple linear regressions is more accurate because it allows for the structural descriptors for each drug-DNA propriety of the 25 molecules to be connected to quantitatively evaluate the effect of the substituent. The selected descriptors are: the lowest unoccupied molecular orbital energy, ELUMO; the electrophilicity index, ω; and the activation energy, Ea.
The QSPR model built using the multiple linear regression (MLR) method is represented by the following equation:(1)
(1)
A higher correlation coefficient, r, and lower mean squared error, MSE, indicate that the model is more reliable. The Fisher F-test is also used. Given that the p-value is much smaller than 0.05, we are taking less than a 0.01% risk in assuming that the null hypothesis is wrong. Therefore, we can conclude, with confidence, that the model brings a significant amount of information.
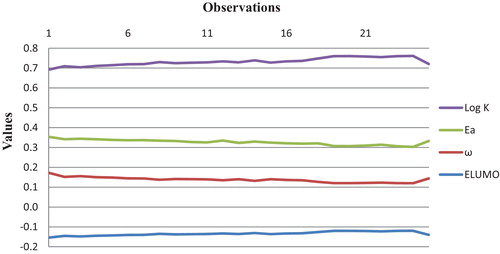
The elaborated QSPR model reveals that the association constant for the drug-DNA could be explained by a number of electronic factors (ELUMO, ω and Ea). The negative correlation of these factors with the association constant for the drug-DNA constants in equation 1 shows that an increase in the values of these factors implies a decrease in the value of Log K, i.e., the variation in Log K with the descriptor values, which are illustrated in , show that the lowest unoccupied molecular orbital, ELUMO, varies in the same way as Log K, so the activation energy and the electrophilicity index vary inversely.
Fig. 2 Variation of the Log K with the selected descriptors by the RLM.
The predicted Log(K) values calculated from Eq. Equation(1)(1)
(1) using the optimal MLR model are given in in comparison to the observed values. The correlation between the predicted and observed Log K (training set and test set) are illustrated in . The descriptors proposed in equation 1 by MLR were therefore used as the input parameters in the multiple non-linear regression (MNLR).
Fig. 3 Correlations of observed and predicted Log K with MLR (training set is in blue and test set is in red).
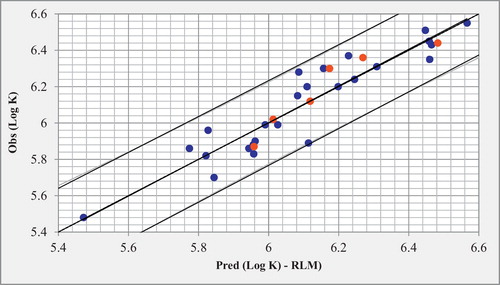
Table 3 The observed, the predicted Log K, and residue according to RLM and RNLM for the 25 acridines derivatives (training set).
3.3 Multiple non-linear regression (MNLR)
We also used the non-linear regression model to improve the structure-propriety relationship to quantitatively evaluate the effect of the substituent. We applied the descriptors proposed by the MLR corresponding to the 25 molecules (training set) to the data matrix. The coefficients, R and R2, were used to select the best regression performance. We used a pre-programmed function of XLSTAT following:where a, b, c, d,… represent the parameters and X1, X2, X3, X4,… represent the variables.
The resulting equation:(2)
(2)
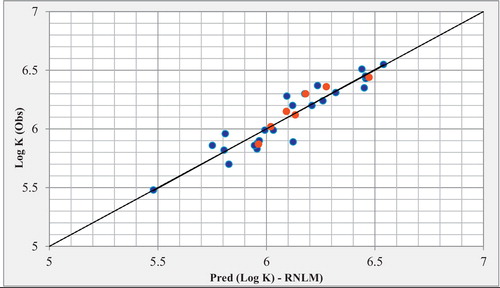
The predicted Log K values calculated from equation 2 are given in in comparison to the observed values. The correlation between the predicted and observed Log K values are shown in .
Fig. 4 Correlations of observed and predicted Log K with MNLR (training set in blue and test set in red).
The true predictive power of a QSPR model is to test their ability to accurately predict the Log K of compounds from an external test set (compounds that were not used for the model development). The Log K values for the remaining set of 6 compounds were deduced from the quantitative model proposed using the 25 molecules (training set) by MLR and MNLR. Their structures are given in ; the observed and calculated log(K) values are given in .
Table 4 The values of the parameters obtained by DFT calculation for the test set compounds.
Table 5 The observed, the predicted Log(K), and residue according to MLR and MNLR for the 6 tested compounds (test set).
A comparison of the log(k-test) to the log(k-obs) values shows that the model made good predictions for the 6 compounds:
From the results obtained by MLR and MNLR, we can conclude that the model performs well, as further supported by the results obtained from testing the 6 test compounds. Even if this good predictive power is the result of chance, we can claim that this is a positive result. Accordingly, this model could be applied to all 9-aniliioacridine derivatives in and add further knowledge to improve the search of antitumour drugs.
A comparison of the quality of the MLR and MNLR models shows that the 2 approaches have a better predictive capability as they give better results. MLR and MNLR were able to establish a satisfactory relationship between the molecular descriptors and the drug-DNA propriety of the studied compounds.
QSPR correlates propriety data with the physicochemical and/or structural properties of a group of compounds. It has been frequently used to predict the proprieties of new compounds and to design compounds with desired properties.
The developed equations can be used for the design of new 9-aniliioacridine derivatives with improved association constants for DNA drug binding properties (log(K)). For example, Eq. Equation(1)(1)
(1) (for RLM) and Eq. Equation(2)
(2)
(2) (for RNLM) indicated the negative correlation of valance first order ELUMO, ω and Ea.
If we develop a new compound with higher values than the existing compounds, it may give rise to the development of more active compounds than those currently in use. In this way, we have designed new compounds () by adding suitable substituents and calculated their propriety using Eqs. Equation(1)(1)
(1) and Equation(2)
(2)
(2) .
Table 6 The proposed novel compounds.
3.4 Proposed novel compounds
The values of the parameters obtained by DFT calculations for the proposed compounds with an association constant for DNA drug binding properties (log(K)) based on the information derived from Eqs. Equation(1)(1)
(1) and Equation(2)
(2)
(2) ().
From the predicted association constant (Log K) for the drug-DNA propriety (), it has been observed that the designed compounds (X13, X14, X15, X16, X17 and X18) have higher Log K values than the existing compounds in the case of the 31 studied compounds (). Additionally, the designed compounds, X1, have lower Log K values than the existing compounds.
4 Conclusion
Multiple linear and non-linear regressions were used to construct a quantitative structure-propriety relation model of 9-aniliioacridine derivatives for their DNA drug binding proprieties. The two regression methods were compared and had a substantially better predictive capability with a greater power. The results show that the models proposed in this paper can predict the association constant for drug-DNA values accurately and that the selected electronic parameters (lowest unoccupied molecular orbital energy, ELUMO, electrophilicity index, ω, and activation energy, Ea), which are sufficiently rich in electronic information to encode structural features, could be used with other descriptors in the development of predictive QSPR models. The accuracy and predictability of the proposed models were illustrated by comparing the key statistical terms r or r2 for the two models (), and the predictive powers of the equations were validated by an external test set ().
We conclude that the most important finding from this research is that we have been able to design and propose new compounds with higher or lower values than existing compounds () by adding suitable substituents by calculating their propriety using the regression equations. Consequently, the proposed models will reduce the time and cost of synthesis as well as the determination of the DNA drug binding capacity of 9-aniliioacridine derivatives.
Acknowledgment
We are grateful to the “Association Marocaine des Chimistes Théoriciens” (AMCT) for its pertinent help concerning the programs.
Notes
Peer review under responsibility of Taibah University.
References
- S.A.GamageD.P.FiggittS.J.WojcikR.K.RalphA.RansijnJ.MauelV.YardleyD.SnowdonS.L.CroftandW.A.DennyJ. Med. Chem.40199726342642
- V.NadarajS.T.SelviS.MohanEur. J. Med. Chem.4432009976980
- S.A.GamageD.P.FiggittS.J.Wojciket alJ. Med. Chem.4016199726342642
- B.F.DickensW.B.WeglickiP.A.BoehmeT.I.MakJ. Mol. Cell. Cardiol.3422002129137
- S.A.GamageN.TepsiriP.Wilairatet alJ. Med. Chem.3710199414861494
- Y.L.ChenC.M.LuI.L.ChenL.T.TsaoJ.P.WangJ. Med. Chem.4521200246894694
- S.M.SondhiM.JoharN.SinghalR.ShuklaR.RaghubirS.G.DastidarIndian J. Chem. B4112200226592666
- I.AntoninCurr. Med. Chem.9200217011716
- M.DemeunynckA.CharmantrayA.MartelliCurr. Pharm. Des.7200117031724
- K.A.WerbovetzP.G.SpoorsR.D.PearsonT.L.MacDonaldMol. Biochem. Parasitol.651994110
- C.S.RouvierJ.M.BarretC.M.FarrellD.SharplesB.T.HillJ.BarbeEur. J. Med. Chem.3912200410291038
- K.RastogiJ.Y.ChangW.Y.Panet alJ. Med. Chem.4520200244854493
- K.M.ChenY.W.SunY.W.TangZ.Y.SunC.H.KwonMol. Pharm.222005118128
- A.AlbertThe Acridines2nd ed.1966Edward ArnoldLondonA.AlbertSelective Toxicity7th ed.1985Chapman & HallLondon
- M.KimuraI.OkabayashiFormation and molecular structure of the novel acridine substituted uracil derivativesJ. Heterocycl. Chem.231986965
- M.DemeunynckF.CharmantrayA.MartelliInterest of acridine derivatives in the anticancer chemotherapyCurr. Pharm. Des.720011703172410.2174/1381612013397131
- M.K.GoftarN.A.RayeniN.MohamadiSpectroscopic studies on the interaction between acridinespermine conjugate with DNAInt. J. Biosci.542014273310.12692/ijb/5.4.27-33
- R.F.LynnetteA.D.WilliamThe genetic toxicology of acridinesMutatton Res.2581991123160
- R.B.SilvermanM.W.HolladayDNA-Interactive Agents, the Organic Chemistry of Drug Design and Drug Action, Chapter 6201410.1016/B978-0-12-382030-3.00006-4
- K.DrlicaR.J.FrancoInhibitors of DNA topoisomerasesBiochemistry277198822532259
- M.J.WaringAnnu. Rev. Biochem.5015901981
- E.M.NelsonK.M.TeweyL.F.LiuProc. Natl. Acad. Sci. U. S. A.8119841361
- M.J.WaringDNA-binding characteristics of acridinylmethanesulfonanilide drugs: comparison with antitumor propertiesEur. J. Cancer1219769951001
- M.KimuraI.OkabayashiJ. Heterocycl. Chem.2331986965967
- M.KimuraA.KatoI.OkabayashiJ. Heterocycl. Chem.29119927380
- M.KimuraQuenching of ethidium-DNA fluorescence by novel acridines with antitumor activitiesYakugaku Zasshi112121992914918
- C.B.BruceA.D.WilliamJ.A.GrahamF.C.BruceJ. Med. Chem.241981170177
- B.C.BaguleyW.A.DennyG.J.AtwellB.F.CainPotential antitumor agents. Quantitative relationships between DNA binding and molecular structure for 9-anflinoacridines substituted in the anilino ringJ. Med. Chem.341981107177
- C.AdamoV.BaroneJ. Chem. Phys. Lett.3302000152M.ParacS.GrimmeJ. Phys. Chem.10620036844Y.YamaguchiS.YokoyamaS.MashikoJ. Chem. Phys.11620026541
- L.BeckerK.HinrichsU.FinkeA new algorithm for computing joins with grid filesProc. of the 9th International Conference on Data EngineeringVienna, Austria1993190197S.J.LeeJ.FinkA.B.BalantekinM.R.StrayerA.S.UmarP.G.ReinhardJ.A.MaruhnW.GreinerPhys. Rev. Lett.601988163
- S.ChtitaM.GhamaliM.LarifA.AdadR.HmammouchiM.BouachrineT.LakhlifiPrediction of biological activity of imidazo[1, 2-a] pyrazine derivatives by combining DFT and QSAR resultsIJIRSET21220137962
- U.SakarR.ParthasarathiV.SubramanianP.K.ChattarajiToxicity analysis of polychlorinated dibenzofurans through globalJ. Mol. IECMD Des.2004124
- XLSTAT 2009 Add-in software (XLSTAT Company). www.xlstat.com.
- S.ChtitaM.LarifM.GhamaliM.BouachrineT.LakhlifiDFT-based QSAR Studies of MK801 derivatives for non competitive antagonists of NMDA using electronic and topological descriptorsJ. Taibah Univ. Sci.201410.1016/j.jtusci.2014.10.006